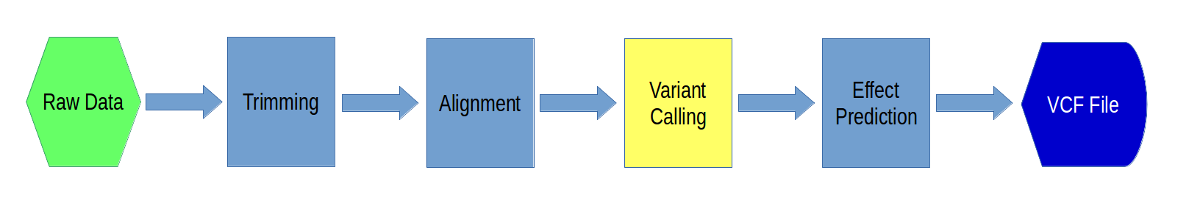
Variant Calling using Freebayes and Delly
Now we will call variants (SNPs and indels) using two different programs, ‘freebayes’ and ‘delly’. We will use the output from the alignment step as input into these programs. In the end you will get a VCF (Variant Call Format) file with genotype information for every variant across all the samples.
1. First, create a new directory for calling variants:
cd ~/variant_example
mkdir 03-freebayes
cd 03-freebayes
Now, let’s link in the relevant files from our alignments, along with their indices:
ln -s ../02-Alignment/*.all.bam .
ln -s ../02-Alignment/*.all.bam.bai .
2. In order to call variants effectively, we need to remove duplicate reads. We will use ‘samtools’ to do this:
module load samtools
samtools rmdup A8100.chr18.all.bam A8100.chr18.all.rmdup.bam
samtools index A8100.chr18.all.rmdup.bam
You will probably get some warning messages… just ignore those. Run ‘samtools rmdup’ and indexing for the rest of the samples as well.
3. Now we will use software called ‘freebayes’ to find SNPs and short indels. Load the module and take a look at the help text:
module load freebayes
freebayes -h
Freebayes has many options, but we are going to run with the defaults. You can set the ploidy as an option, but the default is 2 (i.e. diploid). Also, even with our reduced data set, freebayes will take about 6 hours to run. So, we will run it on the cluster. Download the Slurm script for freebayes:
wget https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2017-August-Variant-Analysis-Workshop/master/wednesday/fb.sh
Change the permissions:
chmod a+x fb.sh
Take a look at the file:
cat fb.sh
The way that we are running freebayes uses, as input, a text file containing a list of BAMs to analyze (the “-L” option). You will need to create this text file before you can run the script:
ls *.all.rmdup.bam > bamlist.txt
Check the file and make sure it looks right:
cat bamlist.txt
There should be five files. Now, run the script using sbatch:
sbatch fb.sh bamlist.txt
4. Now, let’s run delly. We are going to use delly to find large deletions in our data.
module load delly
delly --help
Now, run delly giving it a reference and all of our bam files:
delly -o delly.chr18.all.vcf -g ../ref/chr18.fa *.all.rmdup.bam
This should take about 5 minutes to run.
5. Take a look at the output:
less delly.chr18.all.vcf
We want to just get the variants that “PASS”, not the “LowQual” ones. So we will use a program called ‘awk’ to do that. ‘awk’ is a simple language designed for text processing in Unix. The command below looks at the 7th column ($7) on a line, and if the value of that column is “PASS”, it prints out the line.
grep "^#" delly.chr18.all.vcf > delly.chr18.filtered.vcf
cat delly.chr18.all.vcf | awk '{ if($7=="PASS") print}' >> delly.chr18.filtered.vcf
Take a look at the filtered file. It should only contain “PASS” variants:
less delly.chr18.filtered.vcf
The deletion from the paper is in this file. See if you can find it in the file. Also, look at the VCF specification to get more details of the different fields.