Checking for contaminants
1. Now go back to your ‘rnaseq_example’ directory and create another directory called ‘02-QA’:
cd ~/rnaseq_example
mkdir 02-QA
2. Go into that directory (make sure your prompt shows that you are in the 02-QA directory) and link to all of the files you will be using:
cd 02-QA
ln -s ../00-RawData/*/*.gz .
ls -l
Now you should see a long listing of all the links you just created.
3. Now we want to check for phiX and rRNA contamination. In order to do that we need to download the PhiX genome and Arabidopsis ribosomal RNA. And while we’re at it, let’s download the Arabidopsis genome as well. First, make a directory called ‘ref’ and go into it.
cd ~/rnaseq_example
mkdir ref
cd ref
Then, go to the Illumina iGenomes site. We want to download the PhiX Illumina RTA file to our ‘ref’ directory. In order to do that, we will use the ‘wget’ command. Right click (or whatever is right for your laptop) on the link for PhiX and choose “Copy Link Location” (or something similar). Then use wget to pull down the archive files:
wget ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/PhiX/Illumina/RTA/PhiX_Illumina_RTA.tar.gz
4. Next, we need to uncompress and extract all the files from the archives. We will use the ‘tar’ command. First, let’s take a look at the options of the ‘tar’ command:
man tar
You will see that ‘tar’ has many options… we will be using the “-x”, “-v”, “-z”, and “-f” options, which are used for extraction, verbose information output, unzipping the file, and giving the filename, respectively. Type “q” to exit this screen.
tar -x -v -z -f PhiX_Illumina_RTA.tar.gz
Explore the directory that you’ve just created. You’ll find annotation and indexed sequence for some popular alignment programs.
5. Now, let’s run some Q&A on one pair of our raw data files. First go back to our Q&A directory and load the bowtie2 aligner:
cd ~/rnaseq_example/02-QA
ls -ltrh
module load bowtie2
Take a look at the options to bowtie2:
bowtie2 --help
Notice the Usage that is printed to the screen. We will use the “-p” option to use 8 threads (processors). ‘bt2-idx’ refers to the basename of the index for the genome you are aligning against. First let’s align against PhiX.
bowtie2 -p 8 -x ../ref/PhiX/Illumina/RTA/Sequence/Bowtie2Index/genome -1 C61_S67_L006_R1_001.fastq.gz -2 C61_S67_L006_R2_001.fastq.gz -S phix.sam &> phix.out
We use a relative path to specify the index file, give it the forward and reverse reads for one of the samples, and then redirect the stderr and stdout to a file. This step will take about 10 minutes to run. When it is done take a look at phix.out:
cat phix.out
These are numbers for the alignment percentages of the reads to the genome. “Concordantly” means that the read pair aligned ith the expected relative mate orientation and with the expected range of distances between mates. The numbers should show that most of your reads did NOT align, since this is a measure of PhiX contamination. At the bottom, you get an overall alignment rate, which should be low, i.e. low PhiX contamination.
6. Next we will check for ribosomal RNA contamination. In order to do that, we need to actually get rRNA for Arabidopsis. One way to do that is to go to NCBI and search for them. First, go to NCBI and in the Search dropdown select “Taxonomy” and search for “arabidopsis”.
Click on “Arabidopsis”:
Click on “Arabidopsis” again:
Click on the “Subtree links” for Nucleotide:
Under Molecule Types, click on “rRNA”:
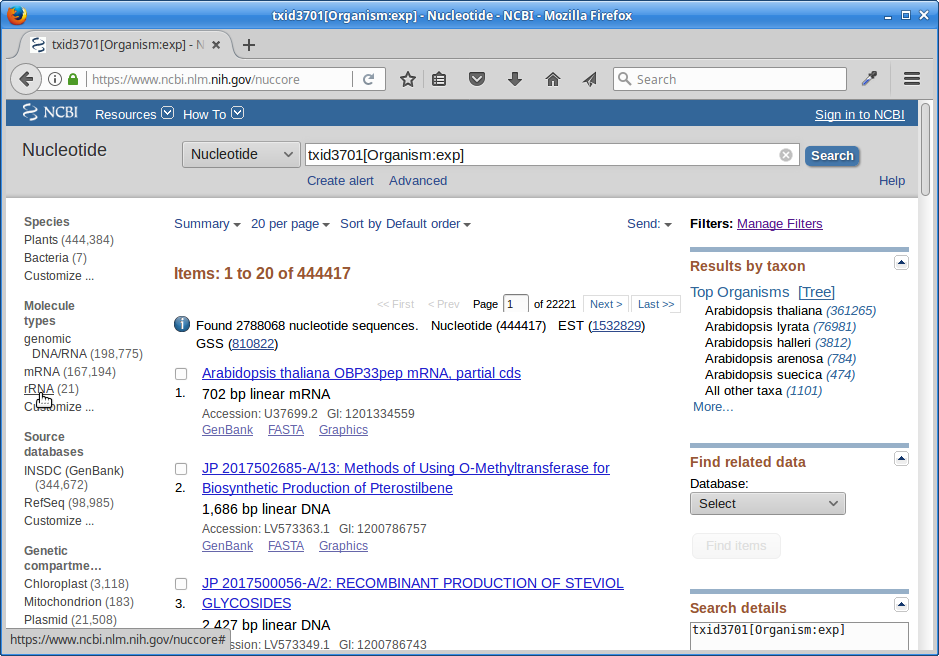
Click on “Send”, choose “File”, choose Format “FASTA”, and click on “Create File”.
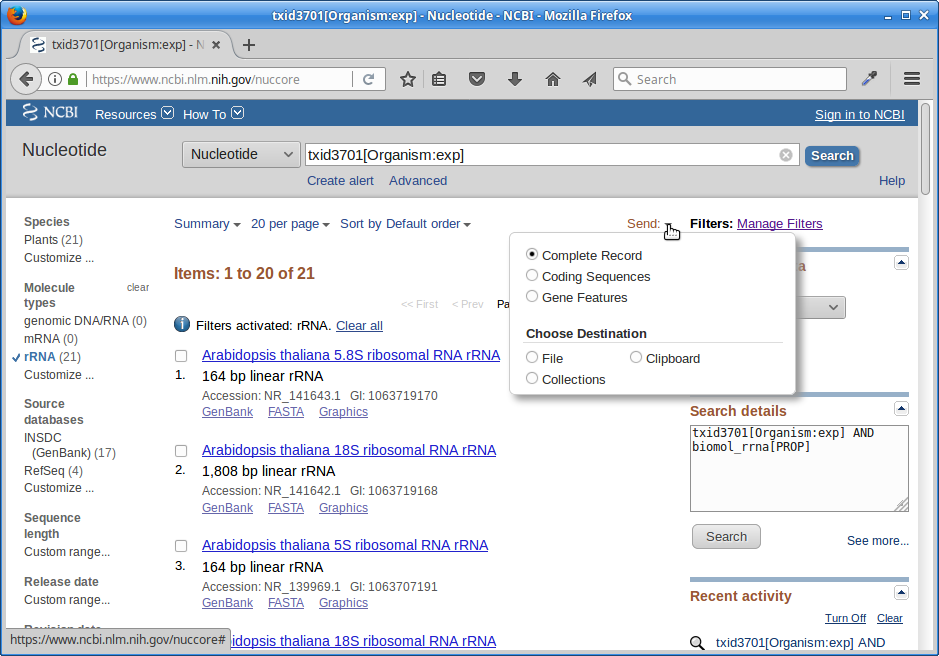
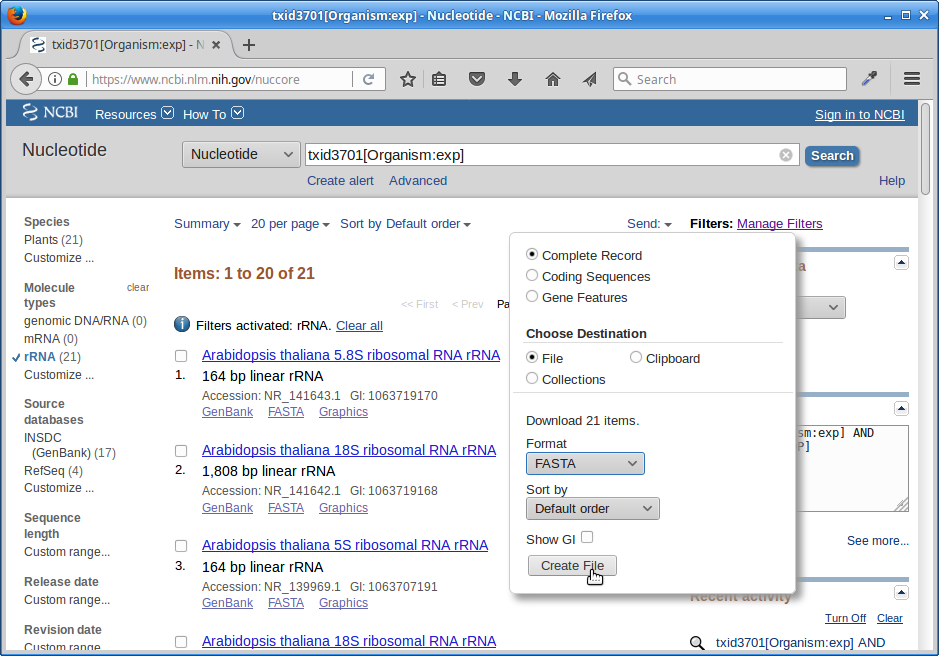
Save this file to your computer, rename it to ‘rrna.fa’, and then upload it to your ref directory on the cluster.
7. Let’s use a different aligner to align to the rRNA sequences. Load the ‘bwa’ module and type ‘bwa’ by itself to get a usage:
module load bwa
bwa
Now type ‘bwa mem’ to get the usage for that sub-command:
bwa mem
‘bwa mem’ does partial alignment, so we generally prefer it over the other bwa alignment methods. In order to align the rRNA sequences, we first need to index them. So go to your ref directory and use the index sub-command to index the rrna.fa file.
cd ~/rnaseq_example/ref
bwa index rrna.fa
Once the indexing is finished, go back to your 02-QA directory and run ‘bwa mem’ using 8 threads, redirecting the sam output to a file:
cd ../02-QA
bwa mem -t 8 ../ref/rrna.fa C61_S67_L006_R1_001.fastq.gz C61_S67_L006_R2_001.fastq.gz > rrna.sam
In order to get alignment information, we have to convert the SAM file to a BAM file using software called ‘samtools’:
module load samtools
samtools view -@ 8 -bS -o rrna.bam rrna.sam
Once that finishes, use the ‘samtools flagstat’ command to see the alignment percentages, and capture the output to a file:
samtools flagstat -@ 8 rrna.bam > rrna.out
Take a look at the file:
cat rrna.out
The relevant number here is the 5th line with a percentage of mapped reads which should, again, be low.