Using the Phyloseq package
The phyloseq package is fast becoming a good way a managing micobial community data, filtering and visualizing that data and performing analysis such as ordination. Along with the standard R environment and packages vegan and vegetarian you can perform virually any analysis. Today we will
- Install R packages 2 Load data straight from dbcAmplicons (biom file)
- Filter out Phylum
- Filter out additional Taxa
- Filter out samples
- Graphical Summaries
- Ordination
- Differential Abundances
installation from bioconductor
We first need to make sure we have the necessary packages: phyloseq, ggplot2, gridExtra, gridR, ape, and edgeR.
#source("http://bioconductor.org/biocLite.R")
#biocLite("phyloseq")
#biocLite("ggplot2")
#biocLite("gridExtra")
#biocLite("edgeR")
#biocLite("vegan")
library(phyloseq)
library(ggplot2)
library(gridExtra)
library(vegan)
## Loading required package: permute
## Loading required package: lattice
## This is vegan 2.4-4
Read in the dataset, biom file generated from dbcAmplicons pipeline
First read in the dataset, see what the objects look like. Our Biom file, produces 3 tables: otu_table, taxa_table, sample_data. Look at the head of each. Get the sample names and tax ranks, finally view the phyloseq object. Lets draw a first bar plot.
slashpile_16sV1V3 <- "16sV1V3.biom"
s16sV1V3 = import_biom(BIOMfilename = slashpile_16sV1V3, parseFunction = parse_taxonomy_default)
colnames(tax_table(s16sV1V3)) <- c("Kingdom", "Phylum", "Class", "Order", "Family", "Genus")
head(otu_table(s16sV1V3))
## OTU Table: [6 taxa and 28 samples]
## taxa are rows
## Slashpile1 Slashpile10 Slashpile11 Slashpile13 Slashpile14
## Taxa_00000 0 0 0 1 1
## Taxa_00001 1 0 0 0 0
## Taxa_00002 2908 1496 110 2870 1761
## Taxa_00003 92 32 6 80 61
## Taxa_00004 336 298 35 414 334
## Taxa_00005 17 5 0 1 6
## Slashpile15 Slashpile16 Slashpile17 Slashpile18 Slashpile19
## Taxa_00000 0 0 0 0 1
## Taxa_00001 0 0 0 0 0
## Taxa_00002 2681 1305 2814 2663 2363
## Taxa_00003 120 12 62 52 80
## Taxa_00004 507 10 205 3 632
## Taxa_00005 5 2 0 0 8
## Slashpile2 Slashpile20 Slashpile21 Slashpile22 Slashpile23
## Taxa_00000 0 0 0 0 0
## Taxa_00001 0 0 0 0 0
## Taxa_00002 1842 1555 1272 2650 2360
## Taxa_00003 38 65 41 104 71
## Taxa_00004 1040 87 242 438 240
## Taxa_00005 0 88 6 4 3
## Slashpile24 Slashpile25 Slashpile26 Slashpile27 Slashpile28
## Taxa_00000 0 0 0 0 0
## Taxa_00001 0 0 0 0 0
## Taxa_00002 3186 3252 1348 1649 3874
## Taxa_00003 105 37 18 84 30
## Taxa_00004 277 9 78 448 7
## Taxa_00005 4 0 3 1 0
## Slashpile3 Slashpile4 Slashpile40 Slashpile5 Slashpile6
## Taxa_00000 2 0 0 1 6
## Taxa_00001 0 0 0 0 0
## Taxa_00002 2687 1234 1462 2518 2163
## Taxa_00003 107 52 58 106 81
## Taxa_00004 476 359 407 428 350
## Taxa_00005 16 7 1 7 37
## Slashpile7 Slashpile8 Slashpile9
## Taxa_00000 0 1 4
## Taxa_00001 0 0 0
## Taxa_00002 4279 2042 2968
## Taxa_00003 120 64 86
## Taxa_00004 369 513 523
## Taxa_00005 1 24 2
head(sample_data(s16sV1V3))
## Depth_cm Dist_from_edge Slash_pile_number primers
## Slashpile1 5 Forest 1 16sV1V3
## Slashpile10 20 15m 2 16sV1V3
## Slashpile11 5 15m 2 16sV1V3
## Slashpile13 5 4.5m 2 16sV1V3
## Slashpile14 20 Edge 2 16sV1V3
## Slashpile15 5 Edge 2 16sV1V3
head(tax_table(s16sV1V3))
## Taxonomy Table: [6 taxa by 6 taxonomic ranks]:
## Kingdom Phylum Class
## Taxa_00000 "d__Archaea" NA NA
## Taxa_00001 "d__Archaea" "p__Crenarchaeota" "c__Thermoprotei"
## Taxa_00002 "d__Bacteria" NA NA
## Taxa_00003 "d__Bacteria" "p__Acidobacteria" NA
## Taxa_00004 "d__Bacteria" "p__Acidobacteria" "c__Acidobacteria_Gp1"
## Taxa_00005 "d__Bacteria" "p__Acidobacteria" "c__Acidobacteria_Gp10"
## Order Family Genus
## Taxa_00000 NA NA NA
## Taxa_00001 "o__Desulfurococcales" "f__Pyrodictiaceae" "g__Pyrolobus"
## Taxa_00002 NA NA NA
## Taxa_00003 NA NA NA
## Taxa_00004 NA NA NA
## Taxa_00005 "o__Gp10" "f__Gp10" "g__Gp10"
rank_names(s16sV1V3)
## [1] "Kingdom" "Phylum" "Class" "Order" "Family" "Genus"
sample_variables(s16sV1V3)
## [1] "Depth_cm" "Dist_from_edge" "Slash_pile_number"
## [4] "primers"
s16sV1V3
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 950 taxa and 28 samples ]
## sample_data() Sample Data: [ 28 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 950 taxa by 6 taxonomic ranks ]
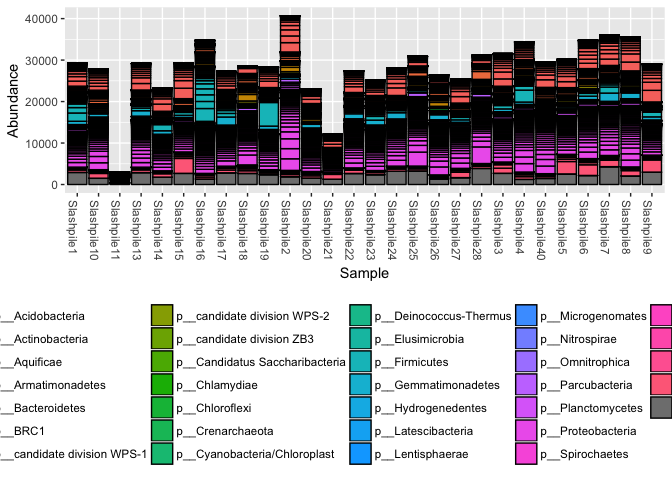
plot_bar(s16sV1V3, fill = "Phylum") + theme(legend.position="bottom")
Filtering our dataset
Lets generate a prevelance table (number of samples each taxa occurs in) for each taxa.
prevelancedf = apply(X = otu_table(s16sV1V3),
MARGIN = 1,
FUN = function(x){sum(x > 0)})
# Add taxonomy and total read counts to this data.frame
prevelancedf = data.frame(Prevalence = prevelancedf,
TotalAbundance = taxa_sums(s16sV1V3),
tax_table(s16sV1V3))
prevelancedf[1:10,]
## Prevalence TotalAbundance Kingdom Phylum
## Taxa_00000 8 17 d__Archaea <NA>
## Taxa_00001 1 1 d__Archaea p__Crenarchaeota
## Taxa_00002 28 63312 d__Bacteria <NA>
## Taxa_00003 28 1864 d__Bacteria p__Acidobacteria
## Taxa_00004 28 9065 d__Bacteria p__Acidobacteria
## Taxa_00005 22 248 d__Bacteria p__Acidobacteria
## Taxa_00006 6 45 d__Bacteria p__Acidobacteria
## Taxa_00007 19 71 d__Bacteria p__Acidobacteria
## Taxa_00008 23 183 d__Bacteria p__Acidobacteria
## Taxa_00009 25 352 d__Bacteria p__Acidobacteria
## Class Order Family
## Taxa_00000 <NA> <NA> <NA>
## Taxa_00001 c__Thermoprotei o__Desulfurococcales f__Pyrodictiaceae
## Taxa_00002 <NA> <NA> <NA>
## Taxa_00003 <NA> <NA> <NA>
## Taxa_00004 c__Acidobacteria_Gp1 <NA> <NA>
## Taxa_00005 c__Acidobacteria_Gp10 o__Gp10 f__Gp10
## Taxa_00006 c__Acidobacteria_Gp11 o__Gp11 f__Gp11
## Taxa_00007 c__Acidobacteria_Gp12 o__Gp12 f__Gp12
## Taxa_00008 c__Acidobacteria_Gp13 o__Gp13 f__Gp13
## Taxa_00009 c__Acidobacteria_Gp15 o__Gp15 f__Gp15
## Genus
## Taxa_00000 <NA>
## Taxa_00001 g__Pyrolobus
## Taxa_00002 <NA>
## Taxa_00003 <NA>
## Taxa_00004 <NA>
## Taxa_00005 g__Gp10
## Taxa_00006 g__Gp11
## Taxa_00007 g__Gp12
## Taxa_00008 g__Gp13
## Taxa_00009 g__Gp15
Whole phylum filtering
First lets remove of the feature with ambiguous phylum annotation.
s16sV1V3.1 <- subset_taxa(s16sV1V3, !is.na(Phylum) & !Phylum %in% c("", "uncharacterized"))
s16sV1V3.1
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 947 taxa and 28 samples ]
## sample_data() Sample Data: [ 28 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 947 taxa by 6 taxonomic ranks ]
Now lets investigate low prevelance/abundance phylum and subset them out.
plyr::ddply(prevelancedf, "Phylum", function(df1){
data.frame(mean_prevalence=mean(df1$Prevalence),total_abundance=sum(df1$TotalAbundance,na.rm = T),stringsAsFactors = F)
})
## Phylum mean_prevalence total_abundance
## 1 p__Acidobacteria 20.463415 143267
## 2 p__Actinobacteria 10.103448 51992
## 3 p__Aquificae 1.333333 4
## 4 p__Armatimonadetes 23.857143 4363
## 5 p__Bacteroidetes 12.171053 35807
## 6 p__BRC1 26.000000 95
## 7 p__candidate division WPS-1 28.000000 6988
## 8 p__candidate division WPS-2 27.000000 1083
## 9 p__candidate division ZB3 1.000000 1
## 10 p__Candidatus Saccharibacteria 28.000000 3335
## 11 p__Chlamydiae 8.800000 81
## 12 p__Chloroflexi 13.444444 6881
## 13 p__Crenarchaeota 1.000000 1
## 14 p__Cyanobacteria/Chloroplast 8.625000 613
## 15 p__Deinococcus-Thermus 3.000000 4
## 16 p__Elusimicrobia 9.333333 50
## 17 p__Firmicutes 8.798246 59168
## 18 p__Gemmatimonadetes 28.000000 22581
## 19 p__Hydrogenedentes 9.000000 16
## 20 p__Latescibacteria 25.000000 398
## 21 p__Lentisphaerae 5.000000 27
## 22 p__Microgenomates 21.000000 192
## 23 p__Nitrospirae 22.000000 1387
## 24 p__Omnitrophica 3.000000 3
## 25 p__Parcubacteria 28.000000 5848
## 26 p__Planctomycetes 21.363636 25294
## 27 p__Proteobacteria 12.359712 303888
## 28 p__Spirochaetes 5.125000 85
## 29 p__SR1 7.000000 15
## 30 p__Tenericutes 1.000000 2
## 31 p__Thermodesulfobacteria 1.000000 1
## 32 p__Verrucomicrobia 19.272727 54748
## 33 <NA> 21.333333 64102
Using the table above, determine the phyla to filter
phyla2Filter = c("p__Aquificae", "p__candidate division ZB3", "p__Crenarchaeota",
"p__Deinococcus-Thermus","p__Omnitrophica","p__Tenericutes",
"p__Thermodesulfobacteria")
# Filter entries with unidentified Phylum.
s16sV1V3.1 = subset_taxa(s16sV1V3.1, !Phylum %in% phyla2Filter)
s16sV1V3.1
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 937 taxa and 28 samples ]
## sample_data() Sample Data: [ 28 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 937 taxa by 6 taxonomic ranks ]
Individual Taxa Filtering
Subset to the remaining phyla by prevelance.
prevelancedf1 = subset(prevelancedf, Phylum %in% get_taxa_unique(s16sV1V3.1, taxonomic.rank = "Phylum"))
ggplot(prevelancedf1, aes(TotalAbundance, Prevalence / nsamples(s16sV1V3.1),color=Phylum)) +
# Include a guess for parameter
geom_hline(yintercept = 0.05, alpha = 0.5, linetype = 2) + geom_point(size = 2, alpha = 0.7) +
scale_x_log10() + xlab("Total Abundance") + ylab("Prevalence [Frac. Samples]") +
facet_wrap(~Phylum) + theme(legend.position="none")
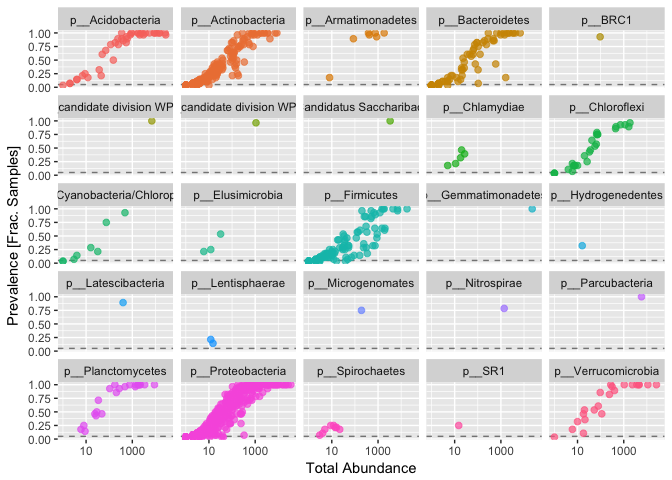
Sometimes you see a clear break, however we aren’t seeing one here. In this case I’m moslty interested in those organisms consistantly present in the dataset, so I’m removing all taxa present in less than 50% of samples.
# Define prevalence threshold as 10% of total samples
prevalenceThreshold = 0.50 * nsamples(s16sV1V3.1)
prevalenceThreshold
## [1] 14
# Execute prevalence filter, using `prune_taxa()` function
keepTaxa = rownames(prevelancedf1)[(prevelancedf1$Prevalence >= prevalenceThreshold)]
length(keepTaxa)
## [1] 381
s16sV1V3.2 = prune_taxa(keepTaxa, s16sV1V3.1)
s16sV1V3.2
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 381 taxa and 28 samples ]
## sample_data() Sample Data: [ 28 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 381 taxa by 6 taxonomic ranks ]
Agglomerate taxa at the Genus level (combine all with the same name) and remove all taxa without genus level assignment
length(get_taxa_unique(s16sV1V3.2, taxonomic.rank = "Genus"))
## [1] 269
s16sV1V3.3 = tax_glom(s16sV1V3.2, "Genus", NArm = TRUE)
s16sV1V3.3
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 268 taxa and 28 samples ]
## sample_data() Sample Data: [ 28 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 268 taxa by 6 taxonomic ranks ]
## out of curiosity how many "reads" does this leave us at???
sum(colSums(otu_table(s16sV1V3.3)))
## [1] 451105
Now lets filter out samples (outliers and low performing samples)
Do some simple ordination looking for outlier samples, first we variance stabilize the data with a log transform, the perform PCoA using bray’s distances
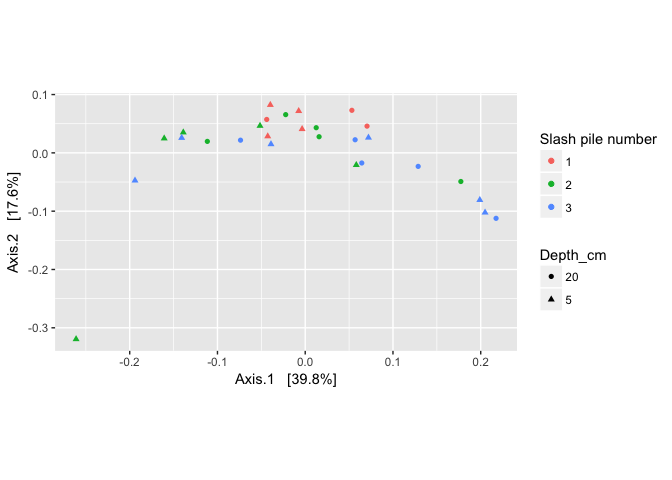
logt = transform_sample_counts(s16sV1V3.3, function(x) log(1 + x) )
out.pcoa.logt <- ordinate(logt, method = "PCoA", distance = "bray")
evals <- out.pcoa.logt$values$Eigenvalues
plot_ordination(logt, out.pcoa.logt, type = "samples",
color = "Slash_pile_number", shape = "Depth_cm") + labs(col = "Slash pile number") +
coord_fixed(sqrt(evals[2] / evals[1]))
plot_ordination(logt, out.pcoa.logt, type = "species", color = "Phylum")
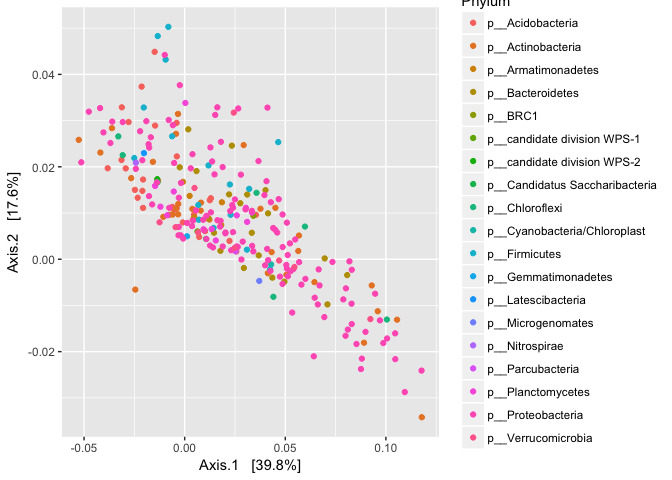
coord_fixed(sqrt(evals[2] / evals[1]))
## <ggproto object: Class CoordFixed, CoordCartesian, Coord>
## aspect: function
## distance: function
## expand: TRUE
## is_linear: function
## labels: function
## limits: list
## range: function
## ratio: 0.664509581087298
## render_axis_h: function
## render_axis_v: function
## render_bg: function
## render_fg: function
## train: function
## transform: function
## super: <ggproto object: Class CoordFixed, CoordCartesian, Coord>
You could also use the MDS method of ordination here, edit the code to do so. Can also edit the distance method used to jaccard, jsd, euclidean. Play with changing those parameters
#Can view the distance method options with
?distanceMethodList
# can veiw the oridinate methods with
?ordinate
Show taxa proportions per sample
grid.arrange(nrow = 2,
qplot(as(otu_table(logt),"matrix")[, "Slashpile18"], geom = "histogram", bins=30) +
xlab("Relative abundance"),
qplot(as(otu_table(logt),"matrix")[, "Slashpile10"], geom = "histogram", bins=30) +
xlab("Relative abundance")
)
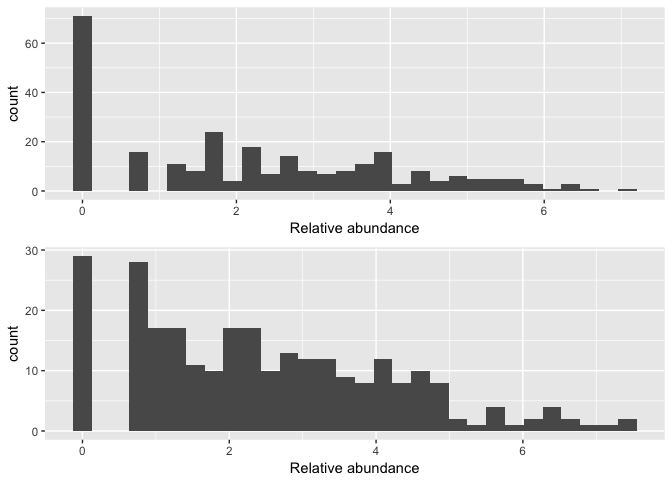
# if you needed to remove candidate outliers, can use the below to remove sample Slashpile18
#s16sV1V3.4 <- prune_samples(sample_names(s16sV1V3.4) != "Slashpile18", s16sV1V3.4)
Look for low perfroming samples
qplot(colSums(otu_table(s16sV1V3.3)),bins=30) +
xlab("Logged counts-per-sample")
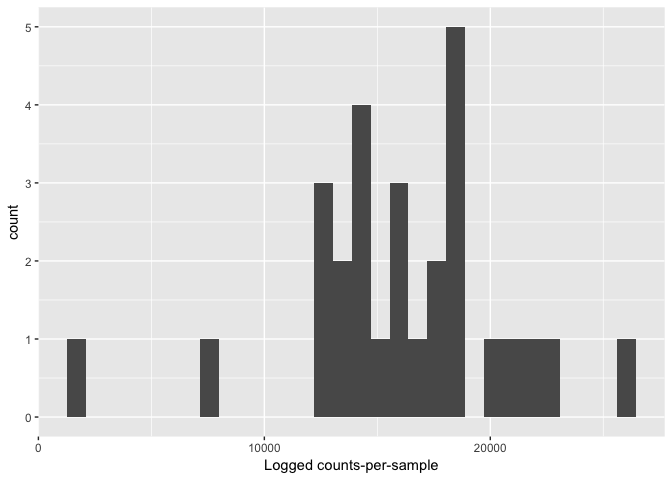
s16sV1V3.4 <- prune_samples(sample_sums(s16sV1V3.3)>=10000, s16sV1V3.3)
s16sV1V3.4
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 268 taxa and 26 samples ]
## sample_data() Sample Data: [ 26 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 268 taxa by 6 taxonomic ranks ]
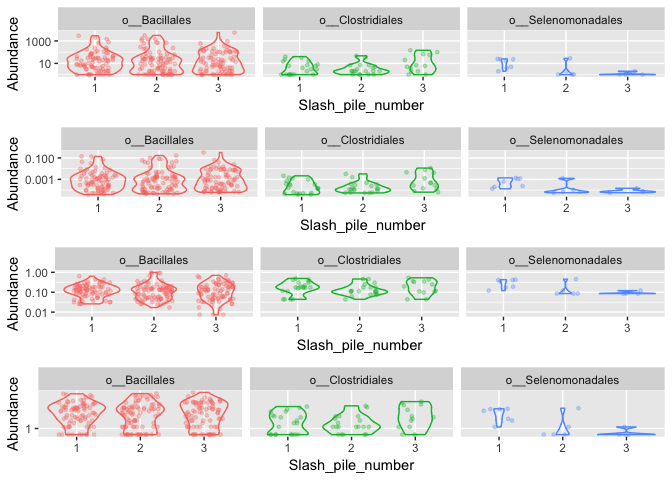
Investigate transformations. We transform microbiome count data to account for differences in library size, variance, scale, etc.
## for Firmictures
plot_abundance = function(physeq, meta, title = "",
Facet = "Order", Color = "Order"){
# Arbitrary subset, based on Phylum, for plotting
p1f = subset_taxa(physeq, Phylum %in% c("p__Firmicutes"))
mphyseq = psmelt(p1f)
mphyseq <- subset(mphyseq, Abundance > 0)
ggplot(data = mphyseq, mapping = aes_string(x = meta,y = "Abundance",
color = Color, fill = Color)) +
geom_violin(fill = NA) +
geom_point(size = 1, alpha = 0.3,
position = position_jitter(width = 0.3)) +
facet_wrap(facets = Facet) + scale_y_log10()+
theme(legend.position="none")
}
# transform counts into "abundances"
s16sV1V3.4ra = transform_sample_counts(s16sV1V3.4, function(x){x / sum(x)})
s16sV1V3.4hell <- s16sV1V3.4
otu_table(s16sV1V3.4hell) <-otu_table(decostand(otu_table(s16sV1V3.4hell), method = "hellinger"), taxa_are_rows=TRUE)
s16sV1V3.4log <- transform_sample_counts(s16sV1V3.4, function(x) log(1 + x))
plotOriginal = plot_abundance(s16sV1V3.4, "Slash_pile_number", title="original")
plotRelative = plot_abundance(s16sV1V3.4ra, "Slash_pile_number", title="relative")
plotHellinger = plot_abundance(s16sV1V3.4hell, "Slash_pile_number", title="Hellinger")
plotLog = plot_abundance(s16sV1V3.4log, "Slash_pile_number", title="Log")
# Combine each plot into one graphic.
grid.arrange(nrow = 4, plotOriginal, plotRelative, plotHellinger, plotLog)
[Normalization and microbial differential abundance strategies depend upon data characteristics] (https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-017-0237-y)
Graphical Summaries
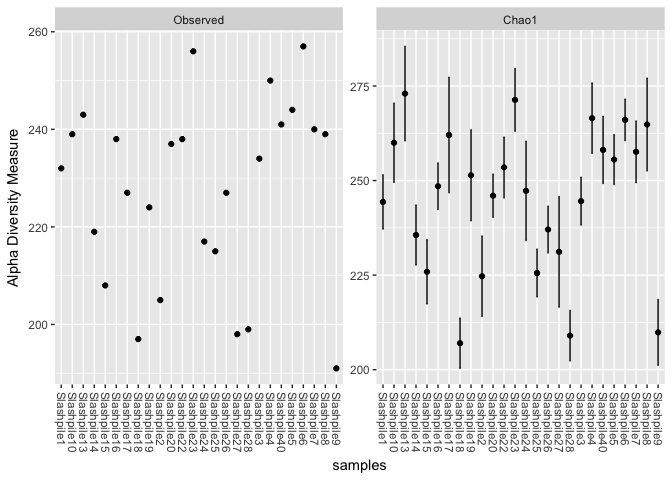
plot_richness(s16sV1V3.4, measures=c("Observed","Chao1"))
## Warning: Removed 26 rows containing missing values (geom_errorbar).
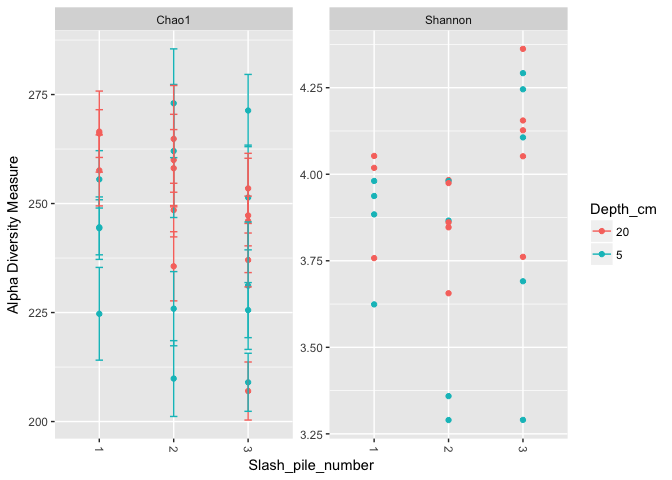
plot_richness(s16sV1V3.4, x = "Slash_pile_number", color="Depth_cm", measures=c("Chao1", "Shannon"))
## Warning: Removed 26 rows containing missing values (geom_errorbar).
# Other Richness measures, "Observed", "Chao1", "ACE", "Shannon", "Simpson", "InvSimpson", "Fisher" try some of these others.
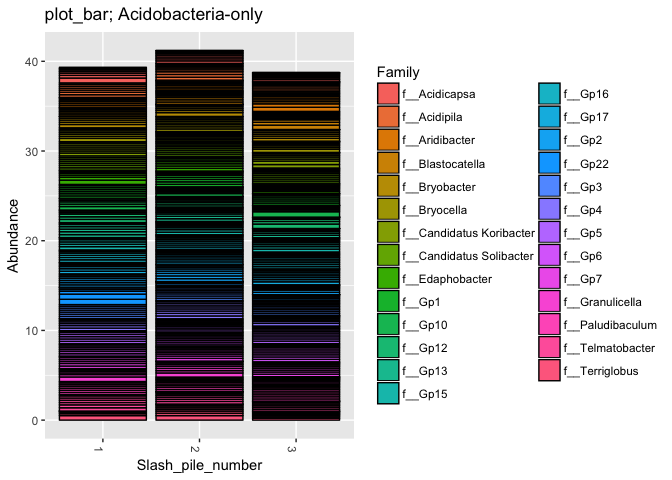
# Subset dataset by phylum
s16sV1V3.4hell_acidob = subset_taxa(s16sV1V3.4hell, Phylum=="p__Acidobacteria")
title = "plot_bar; Acidobacteria-only"
plot_bar(s16sV1V3.4hell_acidob, "Slash_pile_number", "Abundance", "Family", title=title)
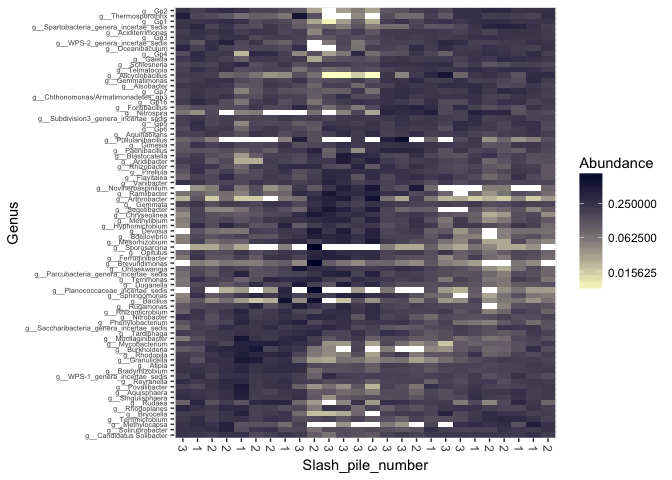
prop = transform_sample_counts(s16sV1V3.4, function(x) x / sum(x) )
keepTaxa <- ((apply(otu_table(prop) >= 0.005,1,sum,na.rm=TRUE) > 2) | (apply(otu_table(prop) >= 0.05, 1, sum,na.rm=TRUE) > 0))
table(keepTaxa)
## keepTaxa
## FALSE TRUE
## 188 80
s16sV1V3.4hell_trim <- prune_taxa(keepTaxa,s16sV1V3.4hell)
plot_heatmap(s16sV1V3.4hell_trim, "PCoA", distance="bray", sample.label="Slash_pile_number", taxa.label="Genus", low="#FFFFCC", high="#000033", na.value="white")
## Warning: Transformation introduced infinite values in discrete y-axis
plot_net(s16sV1V3.4hell_trim, maxdist=0.4, color="Slash_pile_number", shape="Depth_cm")
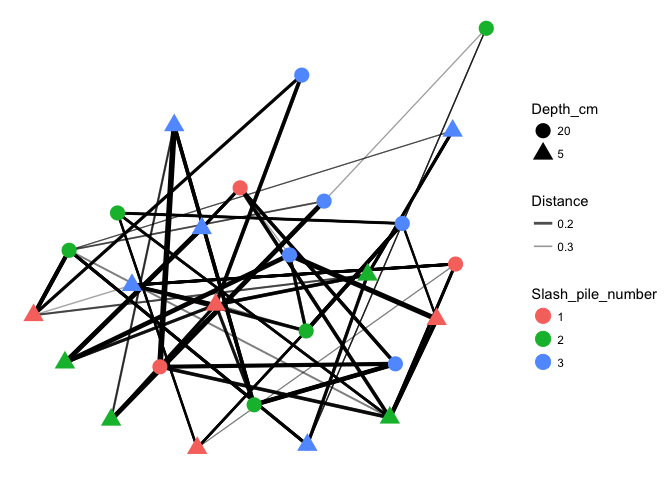
hell.tip.labels <- as(get_variable(s16sV1V3.4hell, "Slash_pile_number"), "character")
# This is the actual hierarchical clustering call, specifying average-linkage clustering
d <- distance(s16sV1V3.4hell, method="bray", type="samples")
hell.hclust <- hclust(d, method="average")
plot(hell.hclust)
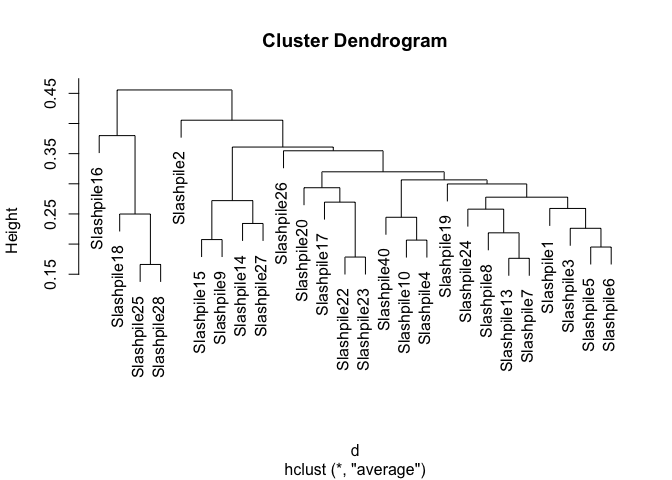
#Lets write out a plot
pdf("My_dendro.pdf", width=7, height=7, pointsize=8)
plot(hell.hclust)
dev.off()
## quartz_off_screen
## 2
png("My_dendro.png", width = 7, height = 7, res=300, units = "in")
plot(hell.hclust)
dev.off()
## quartz_off_screen
## 2
Ordination
v4.hell.ord <- ordinate(s16sV1V3.4hell, "NMDS", "bray")
## Run 0 stress 0.09454798
## Run 1 stress 0.1041802
## Run 2 stress 0.1041538
## Run 3 stress 0.09454804
## ... Procrustes: rmse 1.977178e-05 max resid 4.431723e-05
## ... Similar to previous best
## Run 4 stress 0.1041538
## Run 5 stress 0.09454798
## ... New best solution
## ... Procrustes: rmse 1.507298e-05 max resid 5.150159e-05
## ... Similar to previous best
## Run 6 stress 0.1041808
## Run 7 stress 0.09454799
## ... Procrustes: rmse 1.344327e-05 max resid 3.25311e-05
## ... Similar to previous best
## Run 8 stress 0.1149432
## Run 9 stress 0.1472145
## Run 10 stress 0.1041537
## Run 11 stress 0.1041802
## Run 12 stress 0.1516874
## Run 13 stress 0.09454797
## ... New best solution
## ... Procrustes: rmse 1.539529e-05 max resid 5.360946e-05
## ... Similar to previous best
## Run 14 stress 0.1041539
## Run 15 stress 0.09454797
## ... New best solution
## ... Procrustes: rmse 1.448488e-06 max resid 2.788794e-06
## ... Similar to previous best
## Run 16 stress 0.1502374
## Run 17 stress 0.09454797
## ... Procrustes: rmse 2.484649e-06 max resid 7.468013e-06
## ... Similar to previous best
## Run 18 stress 0.09454797
## ... Procrustes: rmse 4.146237e-06 max resid 1.355354e-05
## ... Similar to previous best
## Run 19 stress 0.09454797
## ... New best solution
## ... Procrustes: rmse 1.465445e-06 max resid 3.117937e-06
## ... Similar to previous best
## Run 20 stress 0.09454797
## ... Procrustes: rmse 2.664022e-06 max resid 8.22876e-06
## ... Similar to previous best
## *** Solution reached
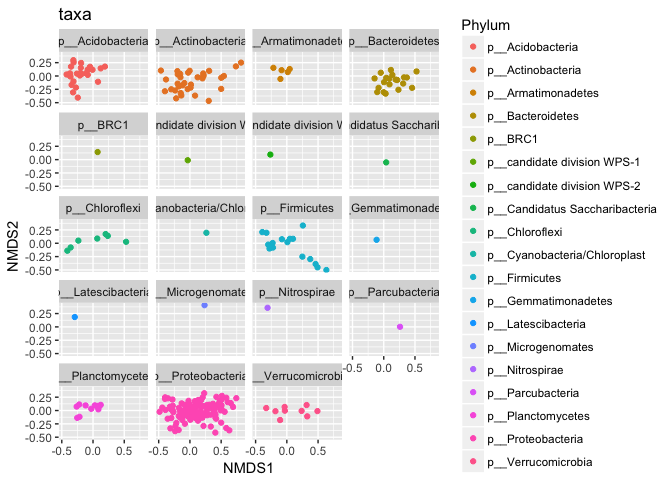
p1 = plot_ordination(s16sV1V3.4hell, v4.hell.ord, type="taxa", color="Phylum", title="taxa")
print(p1)
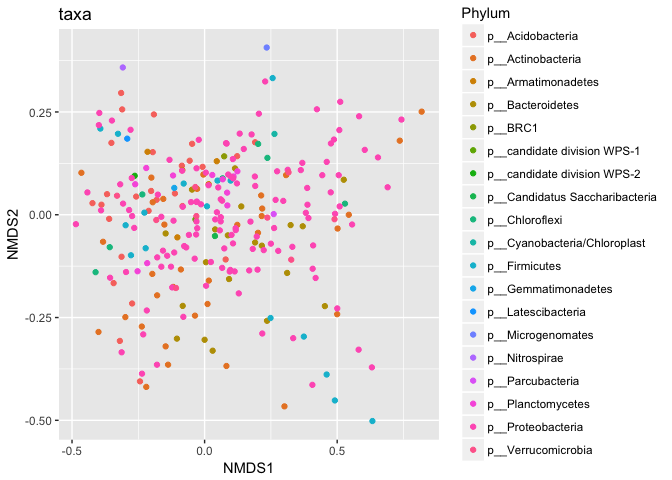
p1 + facet_wrap(~Phylum, 5)
p2 = plot_ordination(s16sV1V3.4hell, v4.hell.ord, type="samples", color="Depth_cm", shape="Slash_pile_number")
p2 + geom_polygon(aes(fill=Slash_pile_number)) + geom_point(size=5) + ggtitle("samples")
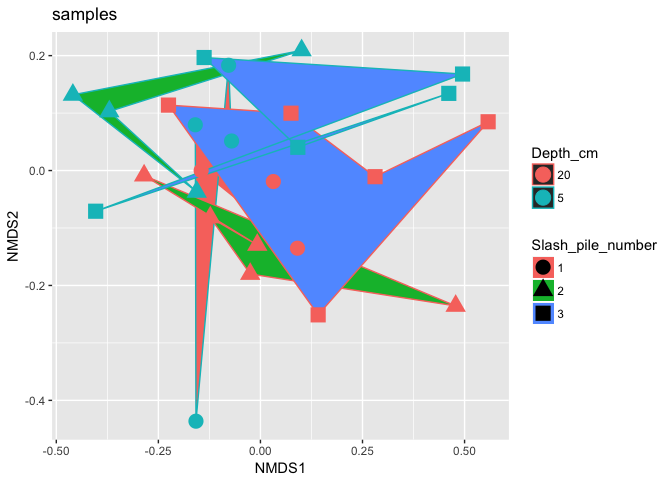
Now try doing oridination with other transformations, such as relative abundance, log. Also looks and see if you can find any trends in the variable Dist_from_edge.
Differential Abundances
For differential abundances we use RNAseq pipeline EdgeR and limma voom.
library("edgeR")
## Loading required package: limma
m = as(otu_table(s16sV1V3.4), "matrix")
# Add one to protect against overflow, log(0) issues.
m = m + 1
# Define gene annotations (`genes`) as tax_table
taxonomy = tax_table(s16sV1V3.4, errorIfNULL=FALSE)
if( !is.null(taxonomy) ){
taxonomy = data.frame(as(taxonomy, "matrix"))
}
# Now turn into a DGEList
d = DGEList(counts=m, genes=taxonomy, remove.zeros = TRUE)
# Calculate the normalization factors
z = calcNormFactors(d, method="RLE")
# Check for division by zero inside `calcNormFactors`
if( !all(is.finite(z$samples$norm.factors)) ){
stop("Something wrong with edgeR::calcNormFactors on this data,
non-finite $norm.factors, consider changing `method` argument")
}
plotMDS(z, col = as.numeric(factor(sample_data(s16sV1V3.4)$Slash_pile_number)), labels = sample_names(s16sV1V3.4))
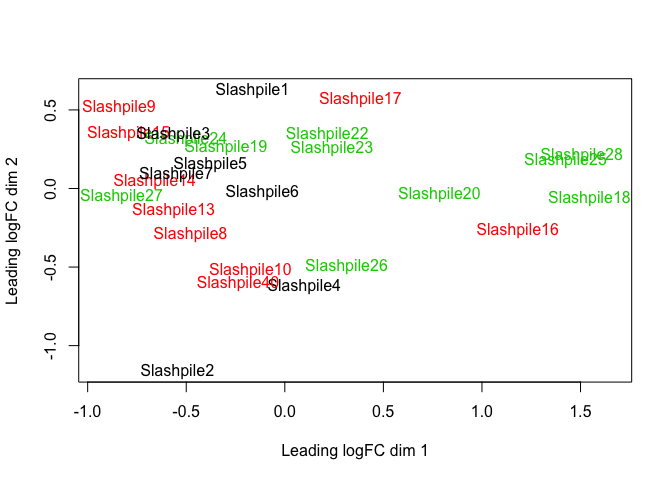
# Creat a model based on Slash_pile_number and depth
mm <- model.matrix(~ Slash_pile_number + Depth_cm, data=data.frame(as(sample_data(s16sV1V3.4),"matrix"))) # specify model with no intercept for easier contrasts
## Warning in class(X) <- NULL: Setting class(x) to NULL; result will no
## longer be an S4 object
mm
## (Intercept) Slash_pile_number2 Slash_pile_number3 Depth_cm5
## Slashpile1 1 0 0 1
## Slashpile10 1 1 0 0
## Slashpile13 1 1 0 1
## Slashpile14 1 1 0 0
## Slashpile15 1 1 0 1
## Slashpile16 1 1 0 0
## Slashpile17 1 1 0 1
## Slashpile18 1 0 1 0
## Slashpile19 1 0 1 1
## Slashpile2 1 0 0 1
## Slashpile20 1 0 1 0
## Slashpile22 1 0 1 0
## Slashpile23 1 0 1 1
## Slashpile24 1 0 1 0
## Slashpile25 1 0 1 1
## Slashpile26 1 0 1 0
## Slashpile27 1 0 1 1
## Slashpile28 1 0 1 1
## Slashpile3 1 0 0 1
## Slashpile4 1 0 0 0
## Slashpile40 1 1 0 0
## Slashpile5 1 0 0 1
## Slashpile6 1 0 0 0
## Slashpile7 1 0 0 0
## Slashpile8 1 1 0 0
## Slashpile9 1 1 0 1
## attr(,"assign")
## [1] 0 1 1 2
## attr(,"contrasts")
## attr(,"contrasts")$Slash_pile_number
## [1] "contr.treatment"
##
## attr(,"contrasts")$Depth_cm
## [1] "contr.treatment"
y <- voom(d, mm, plot = T)
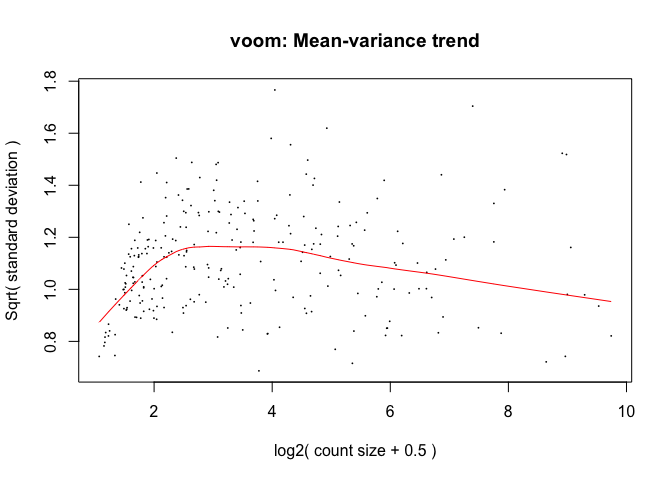
fit <- lmFit(y, mm)
head(coef(fit))
## (Intercept) Slash_pile_number2 Slash_pile_number3 Depth_cm5
## Taxa_00005 9.319727 -0.90802338 -0.5995787 -0.97468059
## Taxa_00007 7.582090 -0.40477165 -0.1401075 0.35726433
## Taxa_00008 8.902649 -0.45194346 -1.0614156 0.14971024
## Taxa_00009 8.520510 0.05426432 0.1214146 1.21637400
## Taxa_00010 11.868568 0.10401232 0.5951760 0.38274218
## Taxa_00011 8.795769 0.29373084 0.4107084 -0.09173845
# Comparison between cultivars C and I5 at time 6
contr <- makeContrasts(Depth5v10 = "Depth_cm5",
levels = colnames(coef(fit)))
## Warning in makeContrasts(Depth5v10 = "Depth_cm5", levels =
## colnames(coef(fit))): Renaming (Intercept) to Intercept
tmp <- contrasts.fit(fit, contr)
## Warning in contrasts.fit(fit, contr): row names of contrasts don't match
## col names of coefficients
tmp <- eBayes(tmp)
tmp2 <- topTable(tmp, coef=1, sort.by = "P", n = Inf)
tmp2$Taxa <- rownames(tmp2)
tmp2 <- tmp2[,c("Taxa","logFC","AveExpr","P.Value","adj.P.Val")]
length(which(tmp2$adj.P.Val < 0.05)) # number of DE genes
## [1] 0
# 0
sigtab = cbind(as(tmp2, "data.frame"), as(tax_table(s16sV1V3.4)[rownames(tmp2), ], "matrix"))
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
sigtabgen = subset(sigtab, !is.na(Genus))
# Phylum order
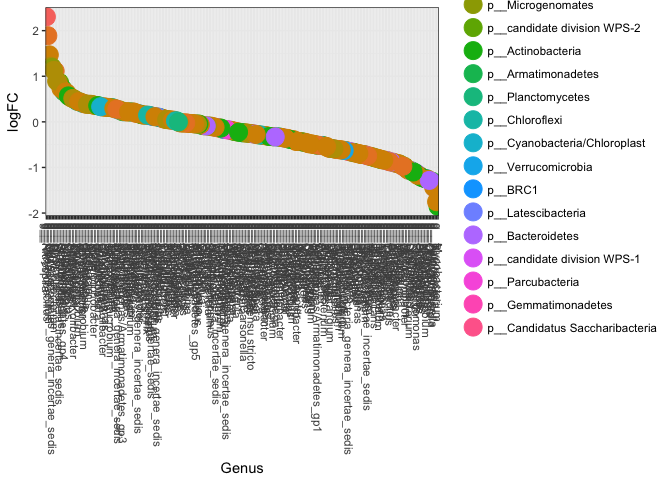
x = tapply(sigtabgen$logFC, sigtabgen$Phylum, function(x) max(x))
x = sort(x, TRUE)
sigtabgen$Phylum = factor(as.character(sigtabgen$Phylum), levels = names(x))
# Genus order
x = tapply(sigtabgen$logFC, sigtabgen$Genus, function(x) max(x))
x = sort(x, TRUE)
sigtabgen$Genus = factor(as.character(sigtabgen$Genus), levels = names(x))
ggplot(sigtabgen, aes(x = Genus, y = logFC, color = Phylum)) + geom_point(size=6) +
theme(axis.text.x = element_text(angle = -90, hjust = 0, vjust = 0.5))