Generating a Gene Expression Matrix
Most analyses have two stages: data reduction and biological analysis. Statistical analyses of scRNA-seq data take as their starting point an expression matrix, where each row represents a gene and each column represents a sample (in scRNAseq a cell). Each entry in the matrix represents the expression level of a particular gene in a given sample (cell). In most cases the number of unique reads assigned to that gene in that sample/cell. Generating the expression matrix often involves some, or all, of the following.
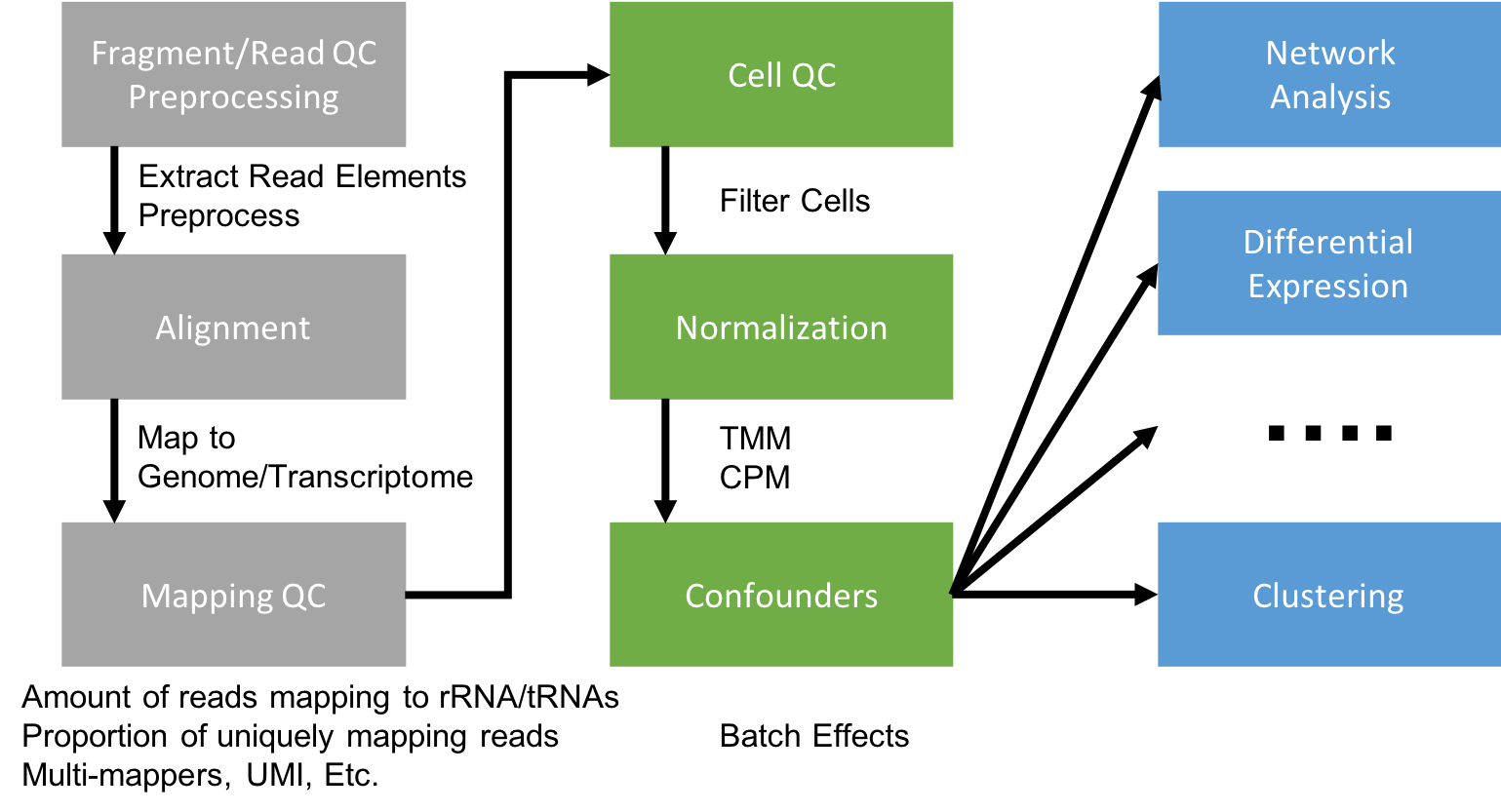
Flowchart of scRNAseq analysis
Preprocessing and mapping reads
Raw fastq files first need to be preprocessed, extracting any elements that are apart of the sequence read.
- Library Barcode (Sample Index) - Used to pool multiple samples on one sequencing lane
- Cell Barcode (10x Barcode) – Used to identify the cell the read came from
- Unique Molecular Index (UMI) – Used to identify reads that arise during PCR replication
- Sequencing Reads – Used to identify the gene a read came from
The remaining sequences can be mapped to a reference genome. We tend to use STAR aligner. For large full-transcript datasets from well annotated organisms (e.g. mouse, human) pseudo-alignment methods (e.g. Kallisto, Salmon) are also a good choice for alignment. For full-length datasets with tens- or hundreds of thousands of reads per cell pseudoaligners become more appealing since their run-time can be several orders of magnitude less than traditional aligners.
Note, if spike-ins are used, the spike-in sequences should be added to the reference sequence prior to mapping.
Mapping QC
After aligning sequence data to the genome we should evaluate the quality of the mapping. There are many ways to measure the mapping quality, including: percentage mapped, amount of reads which map to rRNA/tRNAs, proportion of uniquely mapped reads, reads mapping across splice junctions, read depth along the transcripts, etc. Methods developed for bulk RNAseq, such as RSeQC and samtools, are applicable to single-cell data:
Gene Counting
STAR, Kallisto, and Salmon all quantify the expression level of each gene for
each cell as a part of its output. If UMIs were used, duplicates need to be first marked and then gene expression levels recounted. The packages (UMI-tools)[https://github.com/CGATOxford/UMI-tools) can be used to process and correct UMIs.
Specific steps to be performed are dependant on the type of library, the element layout of the read, and the sequencing parameters.
scRNAseq Libraries
Generating scRNAseq libraries is currently anactive area of research with several protocols being published in the last few years, including:
- CEL-seq Hashimshony, 2012
- CEL-seq2 Hashimshony, 2016
- Drop-seq Macosko, 2015
- InDrop-seq Klein, 2015
- MARS-seq Jaitin, 2014
- SCRB-seq Soumillon, 2014
- Seq-well Gierahn, 2017
- Smart-seq Picelli, 2014
- Smart-seq2 Picelli, 2014
- SMARTer clontech
- STRT-seq Islam, 2014
Differences between the methods are are in how they capture and quantify gene expression (either full-length or tag-based).
Full-length capture tries to achieve a uniform coverage of each transcript (many reads per transcript. Tag-based protocols only capture either the 5’- or 3’-end of each RNA (single read per transcript). Choice in method determines what types of analyses the data can be used for. Full-length capture can be used to distinguise different isoforms, where tag-based method can reduce the mappability and difficult to distinguish different isoforms.
- Tag-based 3’ counting techniques
- 1 read per transcript
- Based on polyA
- Expression analysis only
- Fewer reads per cell needed (~50K reads/cell)
- Less noise in expression patterns
- Full-length
- Based on polyA
- Expression analysis
- Splicing information
- The more information desired beyond expression, the higher the reads needed per cell (~50K reads/cell to 10M reads/cell)
For smaller experiment < 5000 cells, the R packages SingleCellExperiment, scater, SC3 are good choices. For larget experiments (> 5000 cells), the R package Seurat offers a complete solution.
A nice page keeping track of single-cell software can be found here.
10X Genomics generation of expression matrix with cellranger
10X Genomics cell ranger uses the STAR aligner to map reads to a genome after first preprocessing them extracting sample, cell and UMI sequences.
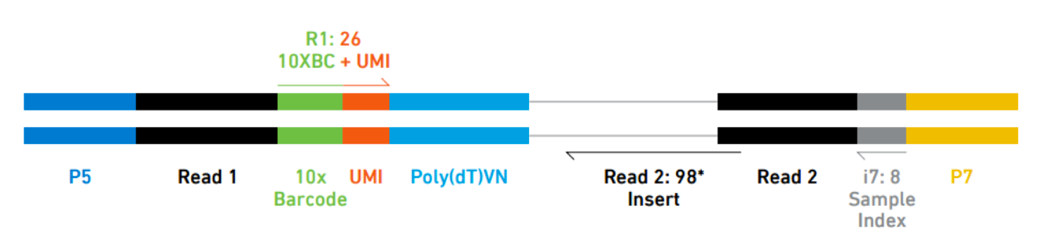
Elements to a 10x read
If using cellranger,
- cellranger mkfastq
- cellranger count
Cell barcode and UMI filtering
- Cell barcodes
- Must be on static list of known cell barcode sequences
- May be 1 mismatch away from the list if the mismatch occurs at a low- quality position (the barcode is then corrected).
- UMIs (Unique Molecular Index)
- Must not be a homopolymer, e.g. AAAAAAAAAA
- Must not contain N
- Must not contain bases with base quality < 10 * UMIs that are 1 mismatch away from a higher-count UMI are corrected to that UMI if they share a cell barcode and gene.
Marking duplicates
- Using only the confidently mapped reads with valid barcodes and UMIs,
*Correct the UMIs
UMIs are corrected to more abundant UMIs that are one mismatch away in sequence.
- Record which reads are duplicates of the same RNA molecule
- Count only the unique UMIs as unique RNA molecules
- These UMI counts form an unfiltered gene-barcode matrix.
Filtering cells (the 10x way)
- Select barcodes that likely contain cells
- Sum UMI counts for each barcode
- Select barcodes with total UMI count >10% of the 99th percentile of the expected recovered cells. ( I honestly haven’t wrapped my head around this yet. )
- Produces a filtered gene-barcode matrix.
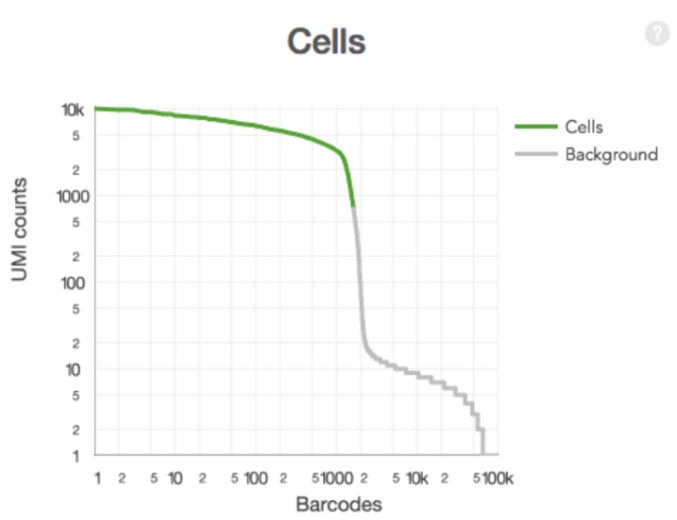
Filtering cells
Marix output
| Type | Description |
|---|---|
| Raw | gene-barcode matrices Contains every barcode from fixed list of known-good barcode sequences. This includes background and non-cellular barcodes. |
| Filtered | gene-barcode matrices Contains only detected cellular barcodes. |
With 3 files needed to completely describe each gene x cell matrix
- matrix.mtx
- genes.tsv
- barcode.tsv
Bam output
10x Chromium cellular and molecular barcode information for each read is stored as TAG fields:
| Tag | Type | Description |
|---|---|---|
| CB | Z | Chromium cellular barcode sequence that is error-corrected and confirmed against a list of known-good barcode sequences. |
| CR | Z | Chromium cellular barcode sequence as reported by the sequencer. |
| CY | Z | Chromium cellular barcode read quality. Phred scores as reported by sequencer. |
| UB | Z | Chromium molecular barcode sequence that is error-corrected among other molecular barcodes with the same cellular barcode and gene alignment. |
| UR | Z | Chromium molecular barcode sequence as reported by the sequencer. |
| UY | Z | Chromium molecular barcode read quality. Phred scores as reported by sequencer. |
| BC | Z | Sample index read. |
| QT | Z | Sample index read quality. Phred scores as reported by sequencer. |
| TR | Z | Trimmed sequence. For the Single Cell 3’ v1 chemistry, this is trailing sequence following the UMI on Read 2. For the Single Cell 3’ v2 chemistry, this is trailing sequence following the cell and molecular barcodes on Read 1. |
| TQ | Z | Trimmed sequence quality. Phred scores as reported by the sequencer. |
The following TAG fields are present if a read maps to the genome and overlaps an exon by at least one base pair. A read may align to multiple transcripts and genes, but it is only considered confidently mapped to the transcriptome it if mapped to a single gene.
| Tag | Type | Description |
|---|---|---|
| TX | Z | Semicolon-separated list of transcripts that are compatible with this alignment. Transcripts are specified with the transcript_id key in the reference GTF attribute column. The format of each entry is [transcript_id],[strand][pos],[cigar], where strand is either + or -, pos is the alignment offset in transcript coordinates, and cigar is the CIGAR string in transcript coordinates. |
| GX | Z | Semicolon-separated list of gene IDs that are compatible with this alignment. Gene IDs are specified with the gene_id key in the reference GTF attribute column. |
| GN | Z | Semicolon-separated list of gene names that are compatible with this alignment. Gene names are specified with gene_name key in the reference GTF attribute column. |
| MM | i | Set to 1 if the genome-aligner (STAR) originally gave a MAPQ < 255 (it multi-mapped to the genome) and Cell Ranger changed it to 255 because the read overlapped exactly one gene. |
| RE | A | Single character indicating the region type of this alignment (E = exonic, N = intronic, I = intergenic). |
An example read
J00113:284:HG27NBBXX:8:2202:16741:8594 1040 1 4491502 255 101M * 0 0 ACTGGACAGTGATTGTGGGGAGCAAGTCCCTCAAGGCATTTAAAACAAAAATCTCGTGTAGCCCCTCAACTGTTCAAGTGGCAGACAAAATAAATTACCAT -A-AAJJJFAFA-F«<JFFJFA-AF)JFFAJJFJAFFA7<JFFJA<JJFA<F<JFJJFAJAJFJFFJFJJJJJJJFJJJJJJFJFJFAFJFJJJJF«<A NH:i:1 HI:i:1 AS:i:99 nM:i:0 TX:Z:ENSMUST00000027035,+2455,101M;ENSMUST00000195555,+1624,101M;ENSMUST00000192650,+3029,101M GX:Z:ENSMUSG00000025902 GN:Z:Sox17 RE:A:E CR:Z:CAAGATCTCGCAAACT CY:Z:AAFFFJJJJJJJJJJJ CB:Z:CAAGATCTCGCAAACT-1 UR:Z:GCCGAGACCT UY:Z:JJJJJJJJJJ UB:Z:GCCGAGACCT BC:Z:CAGCATCA QT:Z:AAFFFFJJ TR:Z:TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTAAAACTACCAGGAGGTATTTCATAGCGAGAAAACACACGCCC TQ:Z:JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ———–7-7<))——–77—))7<AA7A–)))) RG:Z:654:MissingLibrary:1:HG27NBBXX:8
10X genomics sample report
Summary of the alignment and assignment of reads to cells and genes are present in the metrics_summary.csv.
| Metric | Description |
|---|---|
| Estimated Number of Cells | The number of barcodes associated with cell-containing partitions, estimated from the barcode UMI count distribution. |
| Mean Reads per Cell | The total number of sequenced reads divided by the estimated number of cells. |
| Median Genes per Cell | The median number of genes detected (with nonzero UMI counts) across all cell-associated barcodes. |
| Number of Reads | Total number of sequenced reads. |
| Valid Barcodes | Fraction of reads with cell-barcodes that match the whitelist. |
| Reads Mapped Confidently to Transcriptome | Fraction of reads that mapped to a unique gene in the transcriptome with a high mapping quality score as reported by the aligner. |
| Reads Mapped Confidently to Exonic Regions | Fraction of reads that mapped to the exonic regions of the genome with a high mapping quality score as reported by the aligner. |
| Reads Mapped Confidently to Intronic Regions | Fraction of reads that mapped to the intronic regions of the genome with a high mapping quality score as reported by the aligner. |
| Reads Mapped Confidently to Intergenic Regions | Fraction of reads that mapped to the intergenic regions of the genome with a high mapping quality score as reported by the aligner. |
| Sequencing Saturation | The fraction of reads originating from an already-observed UMI. This is a function of library complexity and sequencing depth. More specifically, this is the fraction of confidently mapped, valid cell-barcode, valid UMI reads that had a non-unique (cell-barcode, UMI, gene). This metric was called “cDNA PCR Duplication” in versions of Cell Ranger prior to 1.2. |
| Q30 Bases in Barcode | Fraction of bases with Q-score at least 30 in the cell barcode sequences. This is the i7 index (I1) read for the Single Cell 3’ v1 chemistry and the R1 read for the Single Cell 3’ v2 chemistry. |
| Q30 Bases in RNA Read | Fraction of bases with Q-score at least 30 in the RNA read sequences. This is Illumina R1 for the Single Cell 3’ v1 chemistry and Illumina R2 for the Single Cell 3’ v2 chemistry. |
| Q30 Bases in Sample Index | Fraction of bases with Q-score at least 30 in the sample index sequences. This is the i5 index (I2) read for the Single Cell 3’ v1 chemistry and the i7 index (I1) read for the Single Cell 3’ v2 chemistry. |
| Q30 Bases in UMI | Fraction of bases with Q-score at least 30 in the UMI sequences. This is the R2 read for the Single Cell 3’ v1 chemistry and the R1 read for the Single Cell 3’ v2 chemistry. |
| Fraction Reads in Cells | The fraction of cell-barcoded, confidently mapped reads with cell-associated barcodes. |
| Total Genes Detected | The number of genes with at least one UMI count in any cell. |
| Median UMI Counts per Cell | The median number of total UMI counts across all cell-associated barcodes. |
Exercises
-
Log into ganesh with the username/password given
-
Link the following folder into your home directory
/share/biocore/workshops/2017_2018-single-cell-RNA-sequencing-Workshop-UCD_UCB_UCSF
-
Review cellranger’s sub-applications and help docs
-
Review the 1-mkfastq.slurm script used to generate fastq files from Illumina run output file.
- In the folder 2017_10X_mouse_comparative, which output folders/files were generated from this script?
- Copy over the Reports folder and review it
- Using zless review the first set of reads from sample 654 4. If time remain, mock run the script.
-
Review the 2-counts.slurm script used to map fastq files.
- In the folder 2017_10X_mouse_comparative, which output folders/files were generated from this script?
- Review the metrics_summary.csv file
- What where the total number of reads in this sample?
- Reads Mapped Confidently to Transcriptome?
- Sequencing Saturation?
- Mean Reads per Cell?
- Median UMI Counts per Cell?
- head the files under raw_gene_bc_matrices and filtered_gene_bc_matrices
- If time remain, mock run the script.
- Using samtools and rseqc evaluate the mapping file.
module load rseqc module load samtools bam_stat.py -i possorted_genome_bam.bam samtools flagstat possorted_genome_bam.bam samtools stats possorted_genome_bam.bam
- Run a testrun of cellranger in your home directory
cellranger testrun- view the output
-
Copy the file and extract. Review transfer if needed.
/share/biocore/workshops/2017_2018-single-cell-RNA-sequencing-Workshop-UCD_UCB_UCSF/expression_tables.tar.gz
Commands used in class