Variant Calling (SNPs/INDELs)
This document assumes alignment has been completed.
IF for some reason it didn’t finish, is corrupted or you missed the session, you can copy over from the flash drive
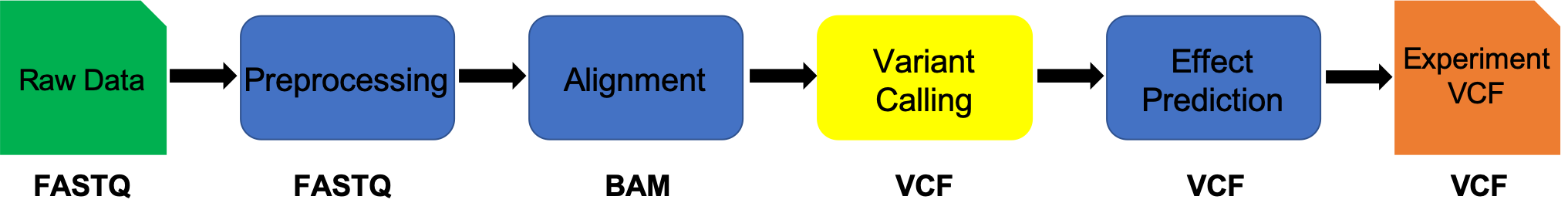
Considerations:
- The quality of the basepair
- The position of the basepair in the read
- The strand of the read
- The mapping quality
Haplotype Calling
Generally speaking there are two popular simple variant (SNPs/INDELs) callers, GATK and Freebayes. Both are now haplotype callers, in the sense that it calls variants based on the literal sequences of reads aligned to the reference region, not their precise alignment (CIGAR alignment). Hapoltype calling avoids one of the core problems with alignment-based variant detection — that identical sequences may have multiple possible alignments:
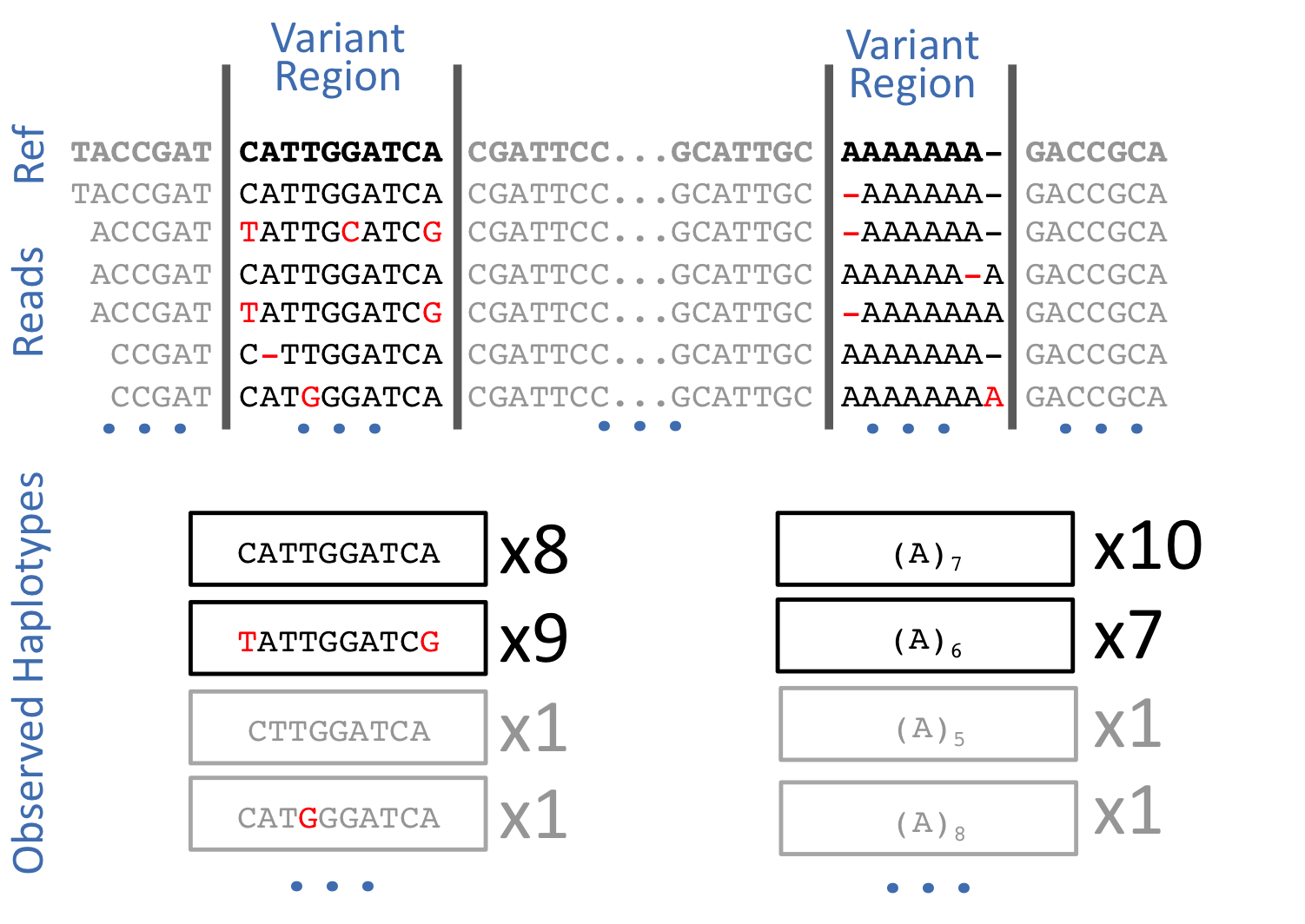
In this workshop we will use Freebayes, which uses short-read alignments for any number of individuals from a population and a reference genome (in FASTA format) to determine the most-likely combination of genotypes for the population at each position in the reference. It reports positions which it finds putatively polymorphic in variant call file (VCF) format.
The definition of a variant is based on the definition of each allele with respect to the reference sequence. We consider 5 major types loosely described as follows.
- SNP The reference and alternate sequences are of length 1 and the base nucleotide is different from one another.
- MNP The reference and alternate sequences are of the same length and have to be greater than 1 and all nucleotides in the sequences differ from one another. OR All reference and alternate sequences have the same length (this is applicable to all alleles).
- INDEL The reference and alternate sequences are not of the same length.
Variant Calling using Freebayes
We will call short variants (SNPs and indels) using freebayes. We will use the output from the prior alignment step as input into the freebayes. freebayes produces a VCF (Variant Call Format) file with genotype information for every variant across all the samples within an experiment.
1. First, lets make sure we are where we are supposed to be:
cd ~/variant_example
2. Now we will use software called ‘freebayes’ to find SNPs and short indels, lets take a look at the help text:
freebayes -h | less
Freebayes has many options, and the help text is long so I’m not going to paste it here but rather we pipe to less to read it.
cd ~/variant_example
curl https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2019-Alliance-for-Global-Health-and-Science-Makerere-University_Variants/master/scripts/freebayes_wks_Variants.sh > freebayes_wks_Variants.sh
cat freebayes_wks_Variant.sh
We are going to run freebayes mostly with the defaults. What non-default parameter do we use and why?
#!/bin/bash
## assumes freebayes is available on the Path
start=`date +%s`
echo $HOSTNAME
outpath='03-Freebayes'
[[ -d ${outpath} ]] || mkdir ${outpath}
outfile=${outpath}/freebayes.vcf
mapfasta=./Reference/GCF_000789395.1_ASM78939v1_genomic.fna
BAMLIST="bamlist.txt"
call="freebayes \
--bam-list ${BAMLIST} \
--fasta-reference ${mapfasta} \
--vcf ${outfile} \
--ploidy 1"
echo $call
eval $call
end=`date +%s`
runtime=$((end-start))
echo $runtime
freebayes can use, as input, a text file containing a list of BAMs to analyze (the “-L” option). You will need to create this text file before you can run the script:
ls 02-BWA/*/*.bam > bamlist.txt
Check the file and make sure it looks right:
cat bamlist.txt
There should be 15 bam files in bamlist.txt. Now, run the script:
bash freebayes_wks_Variants.sh > scriptout/freebayes.out 2> scriptout/freebayes.err
Quality Assurance - bcftools stats.
1. Once the job have finished successfully (check the error and out logs like we did in the previous exercise), then we will run bcltools stats to collect the variant calling stats:
cd ~/variant_example # We'll run this from the main directory
bcftools stats 03-Freebayes/freebayes.vcf > freebayes_stats.txt
less freebayes_stats.txt
plot-vcfstats -p vcfstats freebayes_stats.txt
Scripts
shell script for running freebayes