Variant Annotation (SNPs/INDELs effects)
This document assumes variant calling has been completed.
IF for some reason it didn’t finish, is corrupted or you missed the session, you can copy over from the flash drive
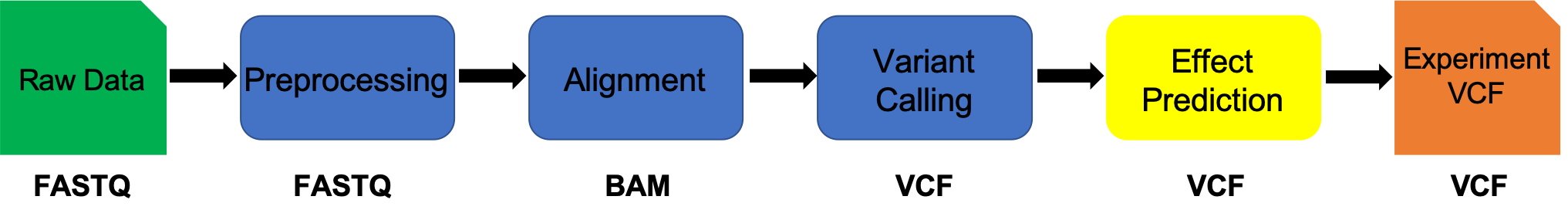
Variant Annotation using SnpEff
We will call annotation our variants (SNPs and indels) using snpeff. We will use the output from the prior variant calling step (vcf) as input into snpeff. snpeff produces a new VCF (Variant Call Format) file with variant annotation added in the INFO field.
1. First, lets make sure we are where we are supposed to be:
cd ~/variant_example
2. We will use software called ‘snpeff’ to annotate the VCF file, lets take a look at the help text:
snpeff -h
SnpEff version SnpEff 4.3t (build 2017-11-24 10:18), by Pablo Cingolani
Usage: snpEff [command] [options] [files]
Run 'java -jar snpEff.jar command' for help on each specific command
Available commands:
[eff|ann] : Annotate variants / calculate effects (you can use either 'ann' or 'eff', they mean the same). Default: ann (no command or 'ann').
build : Build a SnpEff database.
buildNextProt : Build a SnpEff for NextProt (using NextProt\'s XML files).
cds : Compare CDS sequences calculated form a SnpEff database to the one in a FASTA file. Used for checking databases correctness.
closest : Annotate the closest genomic region.
count : Count how many intervals (from a BAM, BED or VCF file) overlap with each genomic interval.
databases : Show currently available databases (from local config file).
download : Download a SnpEff database.
dump : Dump to STDOUT a SnpEff database (mostly used for debugging).
genes2bed : Create a bed file from a genes list.
len : Calculate total genomic length for each marker type.
pdb : Build interaction database (based on PDB data).
protein : Compare protein sequences calculated form a SnpEff database to the one in a FASTA file. Used for checking databases correctness.
seq : Show sequence (from command line) translation.
show : Show a text representation of genes or transcripts coordiantes, DNA sequence and protein sequence.
translocReport : Create a translocations report (from VCF file).
Generic options:
-c , -config : Specify config file
-configOption name=value : Override a config file option
-d , -debug : Debug mode (very verbose).
-dataDir <path> : Override data_dir parameter from config file.
-download : Download a SnpEff database, if not available locally. Default: true
-nodownload : Do not download a SnpEff database, if not available locally.
-h , -help : Show this help and exit
-noLog : Do not report usage statistics to server
-t : Use multiple threads (implies '-noStats'). Default 'off'
-q , -quiet : Quiet mode (do not show any messages or errors)
-v , -verbose : Verbose mode
-version : Show version number and exit
Database options:
-canon : Only use canonical transcripts.
-canonList <file> : Only use canonical transcripts, replace some transcripts using the 'gene_id transcript_id' entries in <file>.
-interaction : Annotate using inteactions (requires interaciton database). Default: true
-interval <file> : Use a custom intervals in TXT/BED/BigBed/VCF/GFF file (you may use this option many times)
-maxTSL <TSL_number> : Only use transcripts having Transcript Support Level lower than <TSL_number>.
-motif : Annotate using motifs (requires Motif database). Default: true
-nextProt : Annotate using NextProt (requires NextProt database).
-noGenome : Do not load any genomic database (e.g. annotate using custom files).
-noExpandIUB : Disable IUB code expansion in input variants
-noInteraction : Disable inteaction annotations
-noMotif : Disable motif annotations.
-noNextProt : Disable NextProt annotations.
-onlyReg : Only use regulation tracks.
-onlyProtein : Only use protein coding transcripts. Default: false
-onlyTr <file.txt> : Only use the transcripts in this file. Format: One transcript ID per line.
-reg <name> : Regulation track to use (this option can be used add several times).
-ss , -spliceSiteSize <int> : Set size for splice sites (donor and acceptor) in bases. Default: 2
-spliceRegionExonSize <int> : Set size for splice site region within exons. Default: 3 bases
-spliceRegionIntronMin <int> : Set minimum number of bases for splice site region within intron. Default: 3 bases
-spliceRegionIntronMax <int> : Set maximum number of bases for splice site region within intron. Default: 8 bases
-strict : Only use 'validated' transcripts (i.e. sequence has been checked). Default: false
-ud , -upDownStreamLen <int> : Set upstream downstream interval length (in bases)
3. First we need to create an annotation database for our organism
cd ~/variant_example/Reference
cp /anaconda3/pkgs/snpeff-4.3.1t-2/share/snpeff-4.3.1t-2/snpEff.config snpEff_edit.config
edit the snpEff_edit.config file using nano, add the genome Clostridium_baratii_str_Sullivan, so that the beginning looks like:
#-------------------------------------------------------------------------------
#
# SnpEff configuration file
#
# Pablo Cingolani
#-------------------------------------------------------------------------------
#---
Clostridium_baratii_str_Sullivan
# Databases are stored here
# E.g.: Information for 'hg19' is stored in data.dir/hg19/
#
# You can use tilde ('~') as first character to refer to your home directory.
# Also, a non-absolute path will be relative to config's file dir
#
#---
data.dir = ./data/
edit the end of the file to include the new database
echo "Clostridium_baratii_str_Sullivan.genome : Clostridium baratii str. Sullivan, complete genome" >> snpEff_edit.config
echo " Clostridium_baratii_str_Sullivan.chromosomes : NZ_CP006905.1, NZ_CP006906.1" >> snpEff_edit.config
echo " Clostridium_baratii_str_Sullivan.NZ_CP006905.1.codonTable : Bacterial_and_Plant_Plastid" >> snpEff_edit.config
echo " Clostridium_baratii_str_Sullivan.NZ_CP006906.1.codonTable : Bacterial_and_Plant_Plastid" >> snpEff_edit.config
We then need to create the proper data directory structure
cd ~/variant_example/Reference
mkdir data
mkdir data/genomes
mkdir data/Clostridium_baratii_str_Sullivan
cp GCF_000789395.1_ASM78939v1_genomic.fna data/genomes/Clostridium_baratii_str_Sullivan.fa
cp GCF_000789395.1_ASM78939v1_genomic.gff data/Clostridium_baratii_str_Sullivan/genes.gff
Finally, build the database
snpeff build -gff3 -c snpEff_edit.config -v Clostridium_baratii_str_Sullivan
finally run snpEff on our vcf file
cd ~/variant_example
snpEff ann -no-downstream -no-upstream -no-utr -o vcf -c Reference/snpEff_edit.config Clostridium_baratii_str_Sullivan 03-Freebayes/freebayes.vcf > 03-Freebayes/freebayes_annotated.vcf