Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: GO.db
Loading required package: AnnotationDbi
Loading required package: stats4
Loading required package: IRanges
Loading required package: S4Vectors
Attaching package: 'S4Vectors'
The following objects are masked from 'package:base':
expand.grid, I, unname
Attaching package: 'IRanges'
The following object is masked from 'package:grDevices':
windows
Loading required package: SparseM
Attaching package: 'SparseM'
The following object is masked from 'package:base':
backsolve
The following object is masked from 'package:IRanges':
members
library(KEGGREST)library(org.Mm.eg.db)
library(pathview)
Pathview is an open source software package distributed under GNU General
Public License version 3 (GPLv3). Details of GPLv3 is available at
http://www.gnu.org/licenses/gpl-3.0.html. Particullary, users are required to
formally cite the original Pathview paper (not just mention it) in publications
or products. For details, do citation("pathview") within R.
The pathview downloads and uses KEGG data. Non-academic uses may require a KEGG
license agreement (details at http://www.kegg.jp/kegg/legal.html).
library(ggplot2)
Files for examples were created in the DE analysis.
Gene Ontology (GO) Enrichment
Gene ontology provides a controlled vocabulary for describing biological processes (BP ontology), molecular functions (MF ontology) and cellular components (CC ontology)
The GO ontologies themselves are organism-independent; terms are associated with genes for a specific organism through direct experimentation or through sequence homology with another organism and its GO annotation.
Terms are related to other terms through parent-child relationships in a directed acylic graph.
Enrichment analysis provides one way of drawing conclusions about a set of differential expression results.
1. topGO Example Using Kolmogorov-Smirnov Testing
Our first example uses Kolmogorov-Smirnov Testing for enrichment testing of our mouse DE results, with GO annotation obtained from the Bioconductor database org.Mm.eg.db.
The first step in each topGO analysis is to create a topGOdata object. This contains the genes, the score for each gene (here we use the p-value from the DE test), the GO terms associated with each gene, and the ontology to be used (here we use the biological process ontology)
topGO by default preferentially tests more specific terms, utilizing the topology of the GO graph. The algorithms used are described in detail here.
head(tab,15)
GO.ID Term
1 GO:0045087 innate immune response
2 GO:0045944 positive regulation of transcription by RNA polymerase II
3 GO:0045766 positive regulation of angiogenesis
4 GO:0045071 negative regulation of viral genome replication
5 GO:0032731 positive regulation of interleukin-1 beta production
6 GO:0051897 positive regulation of protein kinase B signaling
7 GO:0031623 receptor internalization
8 GO:0008360 regulation of cell shape
9 GO:0032760 positive regulation of tumor necrosis factor production
10 GO:0051607 defense response to virus
11 GO:0008150 biological_process
12 GO:0006002 fructose 6-phosphate metabolic process
13 GO:0007229 integrin-mediated signaling pathway
14 GO:0070374 positive regulation of ERK1 and ERK2 cascade
15 GO:0001525 angiogenesis
Annotated Significant Expected raw.p.value
1 530 530 530 4.0e-10
2 768 768 768 3.7e-08
3 97 97 97 2.8e-07
4 49 49 49 4.8e-07
5 51 51 51 1.2e-06
6 61 61 61 1.3e-06
7 79 79 79 1.6e-06
8 105 105 105 1.6e-06
9 85 85 85 1.9e-06
10 228 228 228 2.0e-06
11 10378 10378 10378 2.6e-06
12 10 10 10 3.2e-06
13 63 63 63 6.4e-06
14 108 108 108 6.6e-06
15 300 300 300 7.3e-06
Annotated: number of genes (in our gene list) that are annotated with the term
Significant: n/a for this example, same as Annotated here
Expected: n/a for this example, same as Annotated here
raw.p.value: P-value from Kolomogorov-Smirnov test that DE p-values annotated with the term are smaller (i.e. more significant) than those not annotated with the term.
The Kolmogorov-Smirnov test directly compares two probability distributions based on their maximum distance.
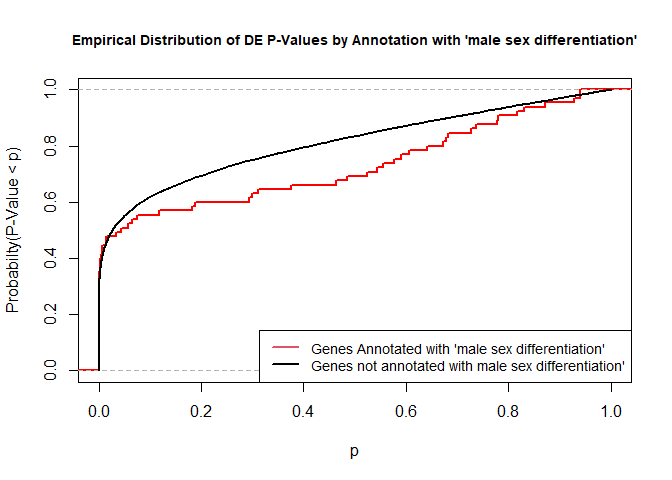
To illustrate the KS test, we plot probability distributions of p-values that are and that are not annotated with the term GO:0046661 “male sex differentiation” (66 genes) p-value 0.8721. (This won’t exactly match what topGO does due to their elimination algorithm):
rna.pp.terms<-genesInTerm(GOdata)[["GO:0046661"]]# get genes associated with termp.values.in<-geneList[names(geneList)%in%rna.pp.terms]p.values.out<-geneList[!(names(geneList)%in%rna.pp.terms)]plot.ecdf(p.values.in,verticals=T,do.points=F,col="red",lwd=2,xlim=c(0,1),main="Empirical Distribution of DE P-Values by Annotation with 'male sex differentiation'",cex.main=0.9,xlab="p",ylab="Probabilty(P-Value < p)")ecdf.out<-ecdf(p.values.out)xx<-unique(sort(c(seq(0,1,length=201),knots(ecdf.out))))lines(xx,ecdf.out(xx),col="black",lwd=2)legend("bottomright",legend=c("Genes Annotated with 'male sex differentiation'","Genes not annotated with male sex differentiation'"),lwd=2,col=2:1,cex=0.9)
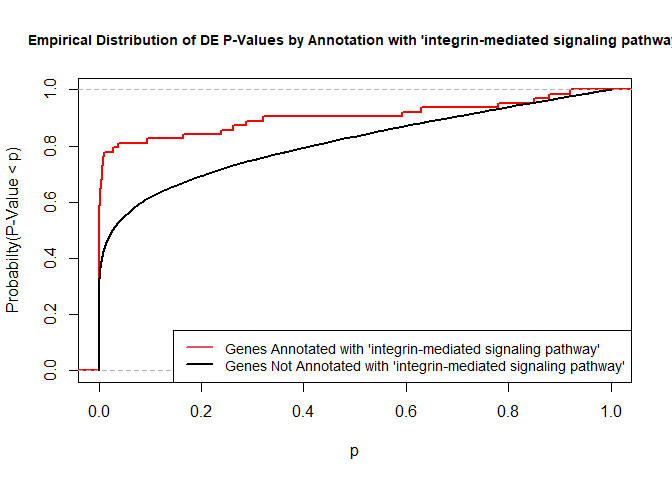
versus the probability distributions of p-values that are and that are not annotated with the term GO:0007229 “integrin-mediated signaling pathway” (66 genes) p-value 9.8x10-5.
rna.pp.terms<-genesInTerm(GOdata)[["GO:0007229"]]# get genes associated with termp.values.in<-geneList[names(geneList)%in%rna.pp.terms]p.values.out<-geneList[!(names(geneList)%in%rna.pp.terms)]plot.ecdf(p.values.in,verticals=T,do.points=F,col="red",lwd=2,xlim=c(0,1),main="Empirical Distribution of DE P-Values by Annotation with 'integrin-mediated signaling pathway'",cex.main=0.9,xlab="p",ylab="Probabilty(P-Value < p)")ecdf.out<-ecdf(p.values.out)xx<-unique(sort(c(seq(0,1,length=201),knots(ecdf.out))))lines(xx,ecdf.out(xx),col="black",lwd=2)legend("bottomright",legend=c("Genes Annotated with 'integrin-mediated signaling pathway'","Genes Not Annotated with 'integrin-mediated signaling pathway'"),lwd=2,col=2:1,cex=0.9)
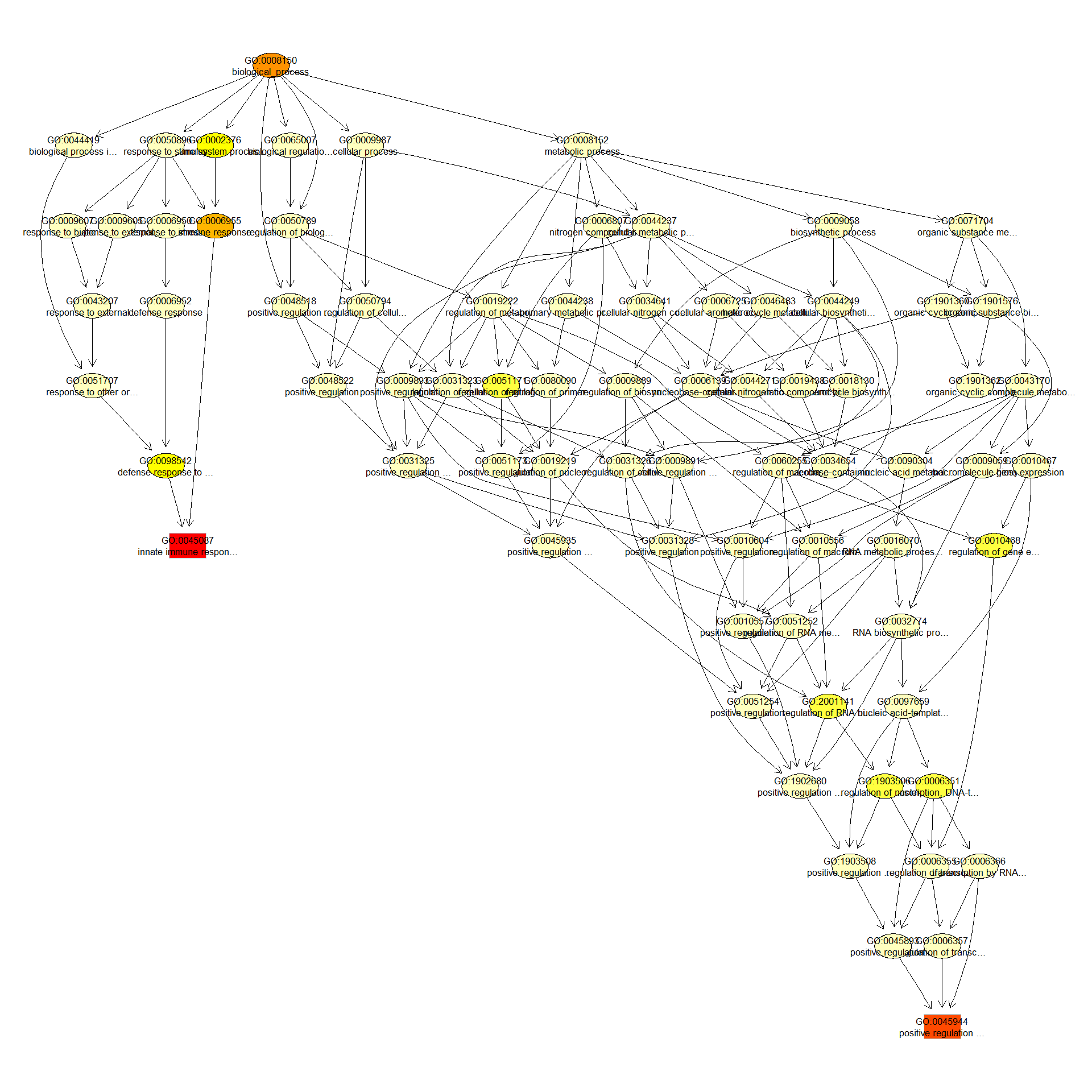
We can use the function showSigOfNodes to plot the GO graph for the 3 most significant terms and their parents, color coded by enrichment p-value (red is most significant):
# Pull all genes for each pathwaypathway.codes<-sub("path:","",names(pathways.list))genes.by.pathway<-sapply(pathway.codes,function(pwid){pw<-keggGet(pwid)if(is.null(pw[[1]]$GENE))return(NA)pw2<-pw[[1]]$GENE[c(TRUE,FALSE)]# may need to modify this to c(FALSE, TRUE) for other organismspw2<-unlist(lapply(strsplit(pw2,split=";",fixed=T),function(x)x[1]))return(pw2)})head(genes.by.pathway)
2. Apply Wilcoxon rank-sum test to each pathway, testing if “in” p-values are smaller than “out” p-values:
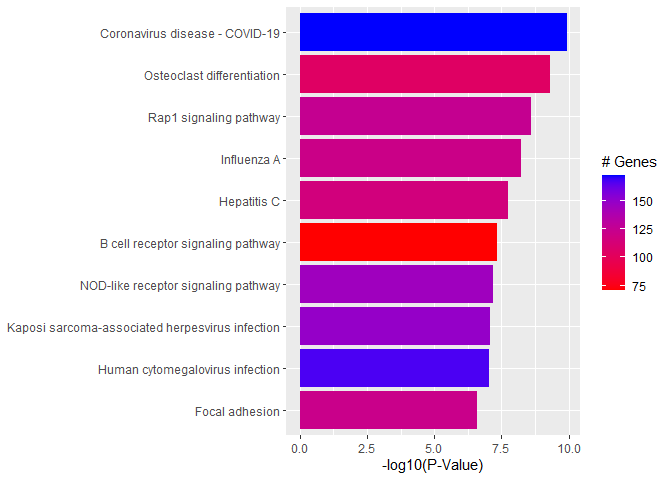
# Wilcoxon test for each pathwaypVals.by.pathway<-t(sapply(names(genes.by.pathway),function(pathway){pathway.genes<-genes.by.pathway[[pathway]]list.genes.in.pathway<-intersect(names(geneList),pathway.genes)list.genes.not.in.pathway<-setdiff(names(geneList),list.genes.in.pathway)scores.in.pathway<-geneList[list.genes.in.pathway]scores.not.in.pathway<-geneList[list.genes.not.in.pathway]if(length(scores.in.pathway)>0){p.value<-wilcox.test(scores.in.pathway,scores.not.in.pathway,alternative="less")$p.value}else{p.value<-NA}return(c(p.value=p.value,Annotated=length(list.genes.in.pathway)))}))# Assemble output tableoutdat<-data.frame(pathway.code=rownames(pVals.by.pathway))outdat$pathway.name<-pathways.list[paste0("path:",outdat$pathway.code)]outdat$p.value<-pVals.by.pathway[,"p.value"]outdat$Annotated<-pVals.by.pathway[,"Annotated"]outdat<-outdat[order(outdat$p.value),]head(outdat)
p.value: P-value for Wilcoxon rank-sum testing, testing that p-values from DE analysis for genes in the pathway are smaller than those not in the pathway
Annotated: Number of genes in the pathway (regardless of DE p-value)
The Wilcoxon rank-sum test is the nonparametric analogue of the two-sample t-test. It compares the ranks of observations in two groups. It is more powerful than the Kolmogorov-Smirnov test.