
Annotating Bacterial Genomes, with a focus on PGAP
Jie Li, PhD, University of California, Davis
The ASM Recording that accompanies this section can be found here
Assess the completeness of the assembled genome
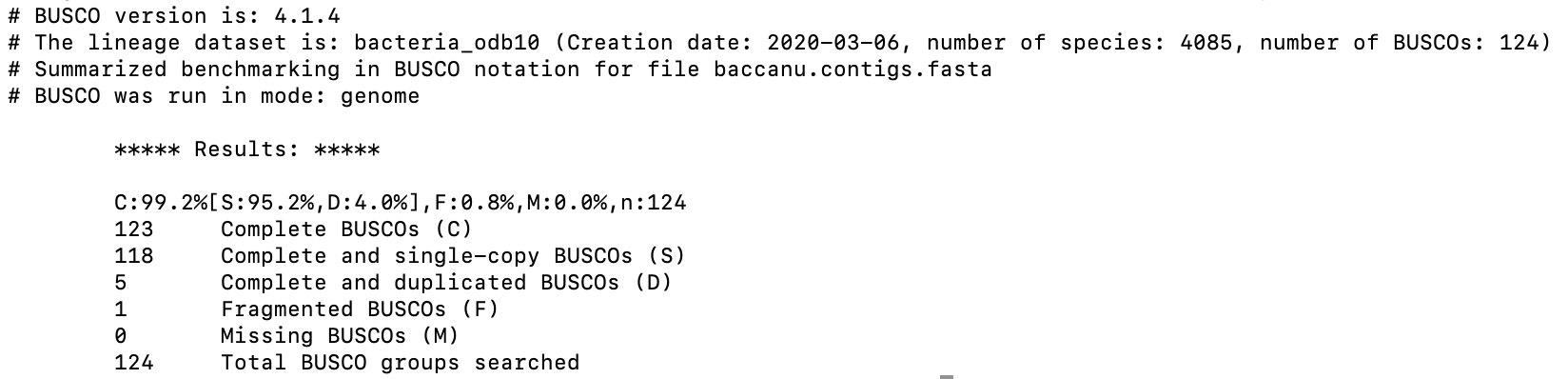
The quality of the assembled genome directly affects the quality of the annotation process. In the previous session, we have assssed the completeness of the assembly by comparing the assembled genome size to the expected genome size. Here, we are going to assess the completeness of the assembly from the gene space point of view. BUSCO will be used for this purpose. The detailed tutorial could be found here. Setting up the configuration file for running BUSCO requires the knowledge of the species that the assembly is for and the domain of species that the assembly should be checked against. Running busco under command line interface is by executing the following command:
busco -i baccanu.contigs.fasta -o canu.busco
The major factors to pay attention to in a BUSCO assessment include: the completeness percentage and the duplication rate. We would like to see as high as possible for the completeness percentage and as low as possible for the duplication rate. The BUSCO result for our assembly is very good.
Introduction to annotation pipelines
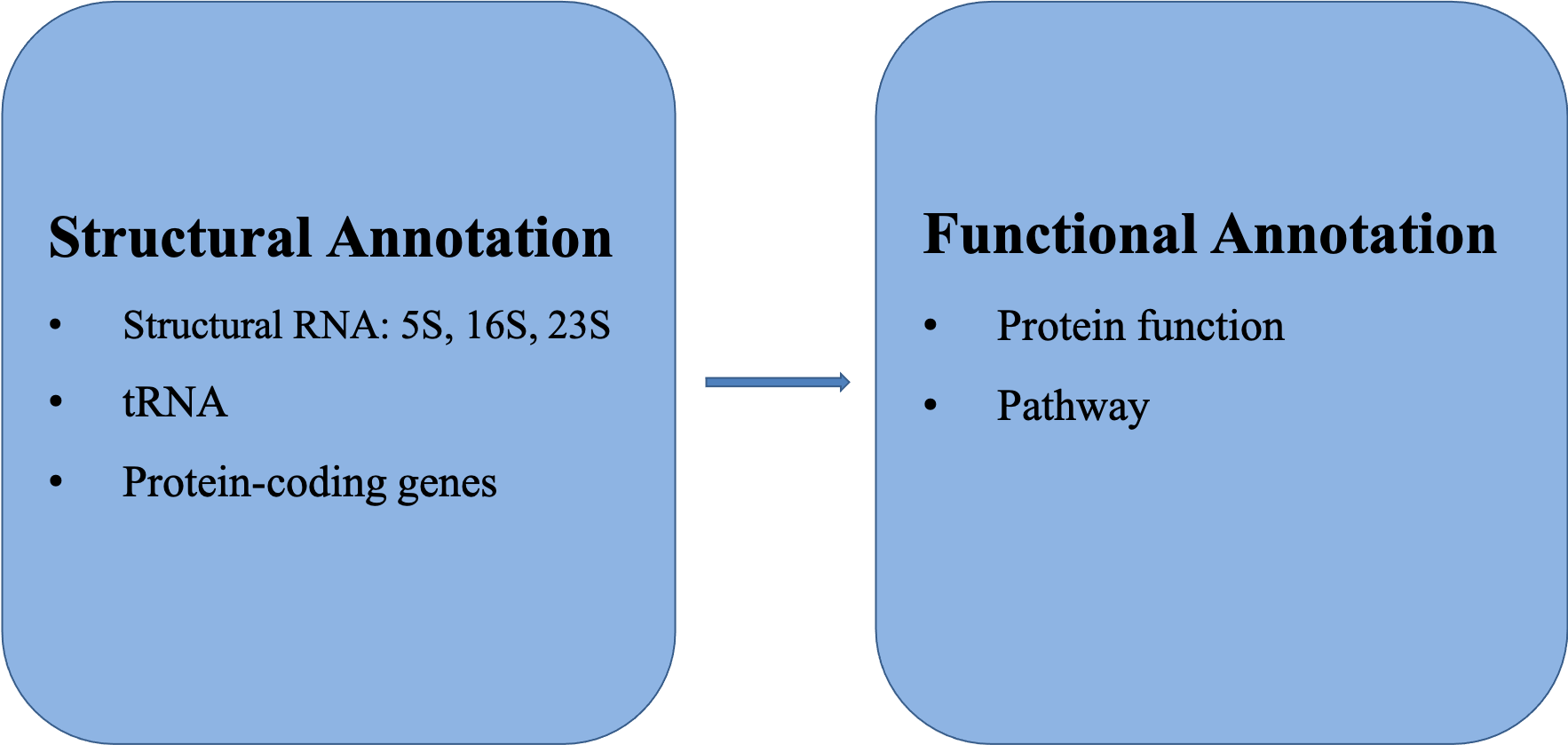
Annotating a bacterial genome involves two major steps: structural annotation and functional annotation. Structural annotation is to identify all relevant genomic sequences for protein coding genes, structural RNA genes, as well as other types of genomic features. The predicted protein coding gene sequences will be used for functional annotation to predict the functions of the proteins, and/or relationship to known pathways.
There are a few annotation pipelines designed for annotating bacterial genomes.
- NCBI’s Prokaryotic Genome Annotation Pipeline PGAP
- Prokka prokarytoic annotation
- RAST Rapid Annotations using Subsystem Technology
- DRAM Distilled and Refined Annotation of Metabolism
These pipelines take the assembled genome fasta file as input. The databases used vary. Prokka and RAST use curated databases, usually smaller, to speed up the searching while maintaining high-quality annotation.
Introduction to PGAP
PGAP annotation for newly assembled bacterial genomes is easy to setup and the requirement in computing resources is reasonable (8 CPUs with 16GB memory or higher, and minimum 80GB disk space). The advantages of using PGAP is not only that it produces NCBI/GenBank recognized file formats, but also the extremely well organized and curated databases used in PGAP.
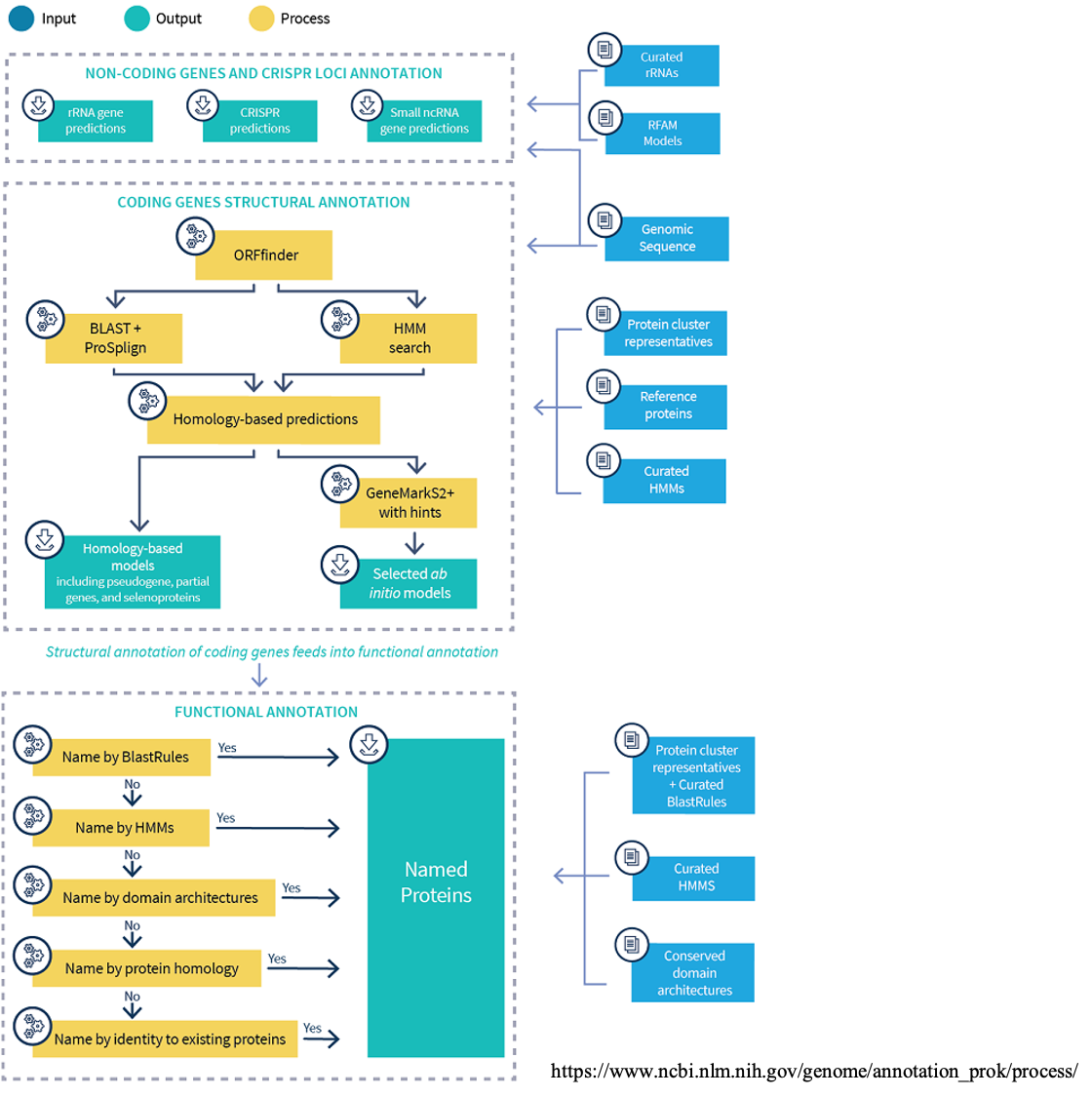
Running PGAP
The computing resources required by PGAP is a machine has 8 CPUs and minimum 2GB memory per CPU. Though I have successfully run PGAP on a laptop that has 4 Cores and 32GB memory. So, the number of CPUs could be lower than 8, but the memory per CPU should be sufficient. Otherwise, one important step in the pipeline (blastp) will fail. PGAP can be run quite easily using docker. The first step in setting up the environment for running PGAP is to install docker on your laptop, or on an AWS instance. Simply download the proper version of docker. The second package required for PGAP is python3.6 or higher. This could be easily installed by installing Anaconda on your machine.
For those who have chosen to use an AWS instance that is Ubuntu platform, after installing docker following the instruction on the docker website, please execute the following commands so that the docker engine is avaible for PGAP scripts.
sudo groupadd docker
sudo usermod -aG docker ${USER}
Then please logout of the AWS instance and re-login to have the group membership re-evaluated.
In order to check whether docker is available, please execute the following command:
docker run hello-world
If this command has executed successfully, you should see a message stating that the “installation appears to be working correctly”.
Once the above two components are properly installed, one could follow the instructions below to obtain PGAP, as well as the most up-to-date databases.
curl -OL https://github.com/ncbi/pgap/raw/prod/scripts/pgap.py
./pgap.py --update
The input to PGAP is the final assembled genome fasta file. One could download the file using the following command if the previous assembly step has not finished.
wget https://bioshare.bioinformatics.ucdavis.edu/bioshare/download/ua4gjm2jyj12oxl/baccanu.contigs.fasta
For those who have Windowns machines, the file could be found here. Please right click on the file baccanu.contigs.fasta and save it to your working directory.
There are two YAML files: input.yaml and submol.yaml, that are required by PGAP for metadata and pipeline parameters. You can download them here the same way as you have downloaded the fasta file above.
Now we are ready to run PGAP. The following command will run the full PGAP pipeline.
./pgap.py -r -o baccanu_anno ./input.yaml
This simply command runs the full PGAP annotation pipeline, in a couple hours on an AWS instance with 8 CPUs and 32GB memory. The results are stored inside baccanu_anno directory. The file annot.gbk is the annotation result in genbank format. The file annot.gff is the annotation result in gff3 format. The file annot.sqn could be submitted to genbank as the included annotation when one submits the assembled genome.