
Multiome profiling with 10X
10X multiome ATAC + Gene Expression kit provides genome-wide profiling of chromatin accessibilities and transcriptome-wide profiling of gene expression at single cell resolution.
Library preparation
10X multiome library preparation proceeds in 4 stages:
- single nuclei isolation Tissues undergo nuclei isolation using Chromium Nuclei Isolation Kit (optimized for frozen tissues) or other nuclei isolation protocols. Please refer to 10X documentation for guidance, cell lines/PBMCs, complex tissues.
- Tn5 Transposation Tn5 transposase mix is applied to the single nuclei suspension created in the first step to fragment the DNA in the open chromatin regions.
- GEM generation and barcoding Transposed single nuclei suspension are partitioned in emulsion oil that contains gel beads inside Chromium Controller. Within each GEM, DNA and RNA fragments are captured and barcoded. Afterwards, the barcoded DNA and RNA fragments are pre-amplified and splitted to ATAC and gene expression libraries.
- Library construction ATAC and gene expression libraries are generated following separate protocols and sequenced.
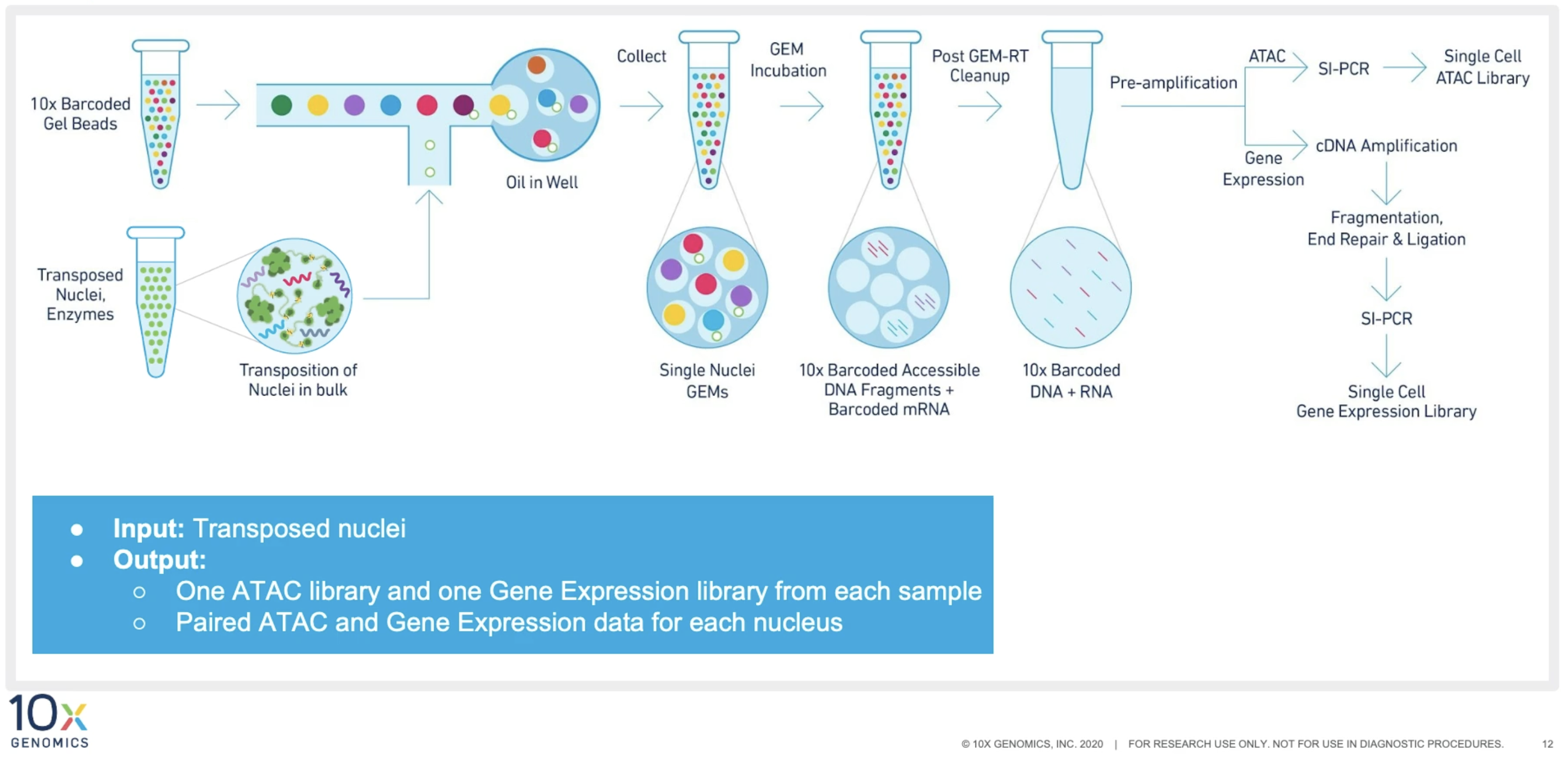
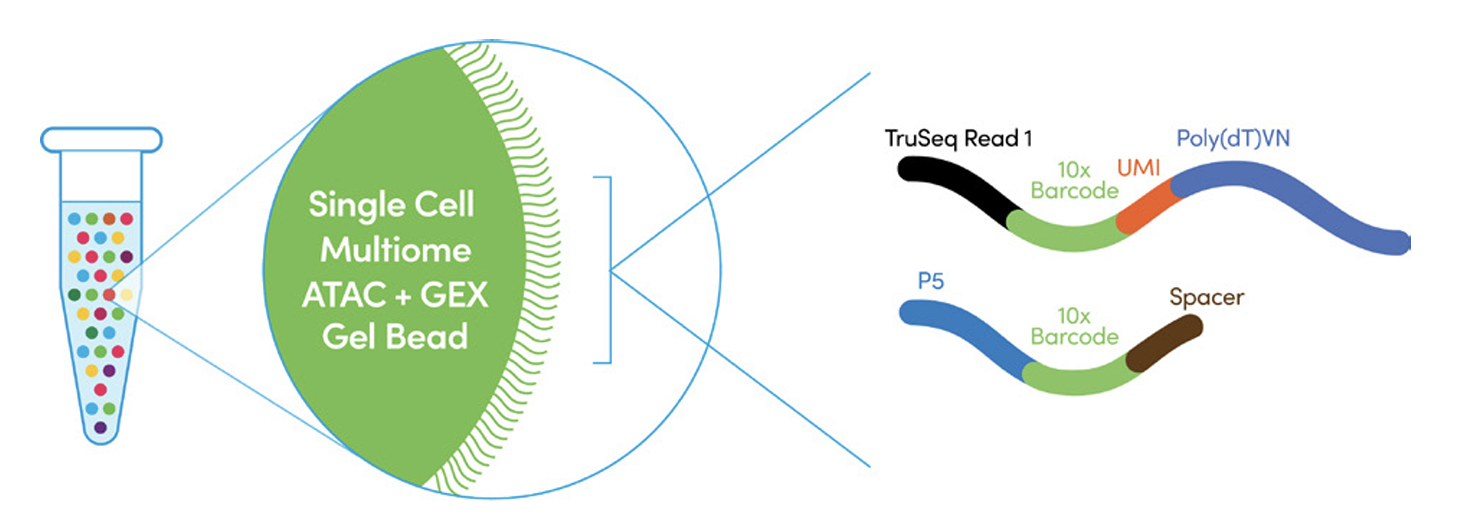
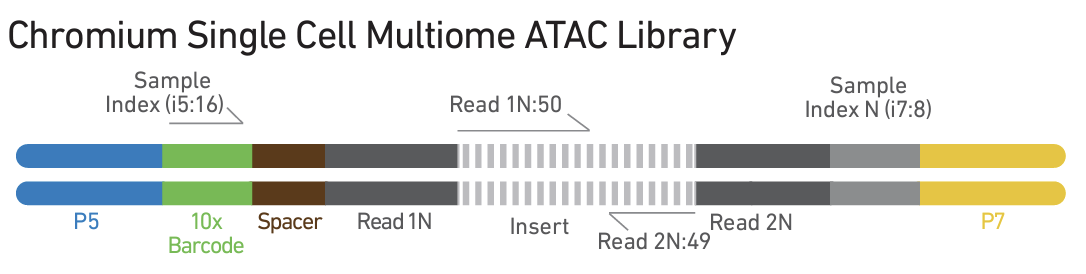
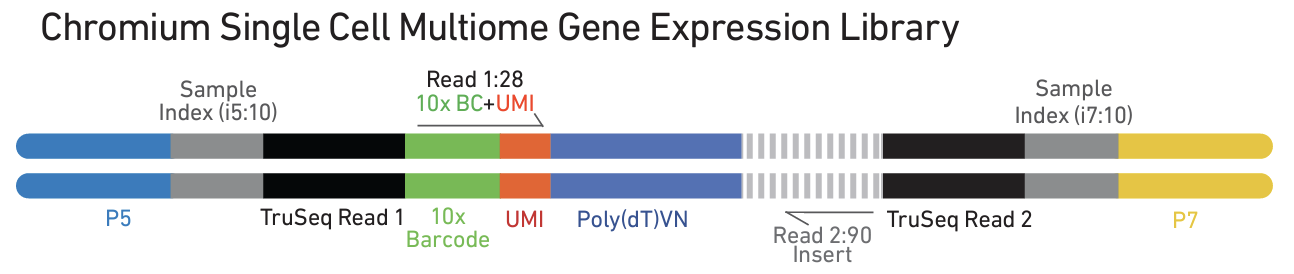
The recommended sequencing depth is 25K/nucleus for ATAC libraries, and 20K/nucleus for gene expression libraries.
Chromatin accessibility profiling with 10X
10x Genomics provides genome-wide profiling of chromatin accessibilities at single cell resolution.
Library preparation
scATAC library preparation occurs in five steps:
- single nuclei isolation Tissues undergo nuclei isolation using Chromium Nuclei Isolation Kit (optimized for frozen tissues) or other nuclei isolation protocols.
- Tn5 Transposation Tn5 transposase mix is applied to the single nuclei suspension created in the first step to fragment the DNA in the open chromatin regions and add Illumina read primer sequences.
- GEM generation and barcoding Transposed single nuclei suspension are partitioned in gel beads, then lysed. DNA fragments are captured and a sequencing primer P5 and a 16 nt 10x (cell) Barcode are added to each fragment. In the same reaction, fragments are amplified.
- GEM pooling and cleanup The DNA from all the gel beads is pooled, and leftover reagents and primers are removed with a magnetic bead purification.
- Library construction Illumina sequencing primer P7 and sample index are added. Completed libraries have both P5 and P7, and are ready for sequencing.
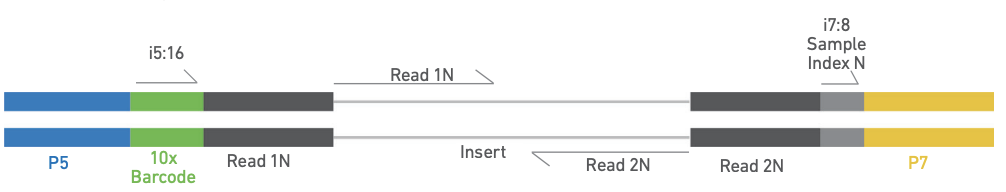
The scATACSeq library
The recommended sequencing depth for single cell ATAC libraries is 25K reads per cell, three orders of magnitude less than the recommended sequencing depth for bulk ATAC libraries.
Example data set for cellranger processing
The data set we will be using in this workshop is an example dataset from 10X
In this study, single nuclei transcriptome and chromatin accessibility profiles were generated from a patient diagnosed with diffuse small lymphocytic lymphoma of the lymph node. The nucleus sequenced are from intra-abdominal lymph node tumor.
For the purposes of this workshop, we are using a subset of this data for data reduction. The full dataset will be used at the data analysis stage.
Workflow
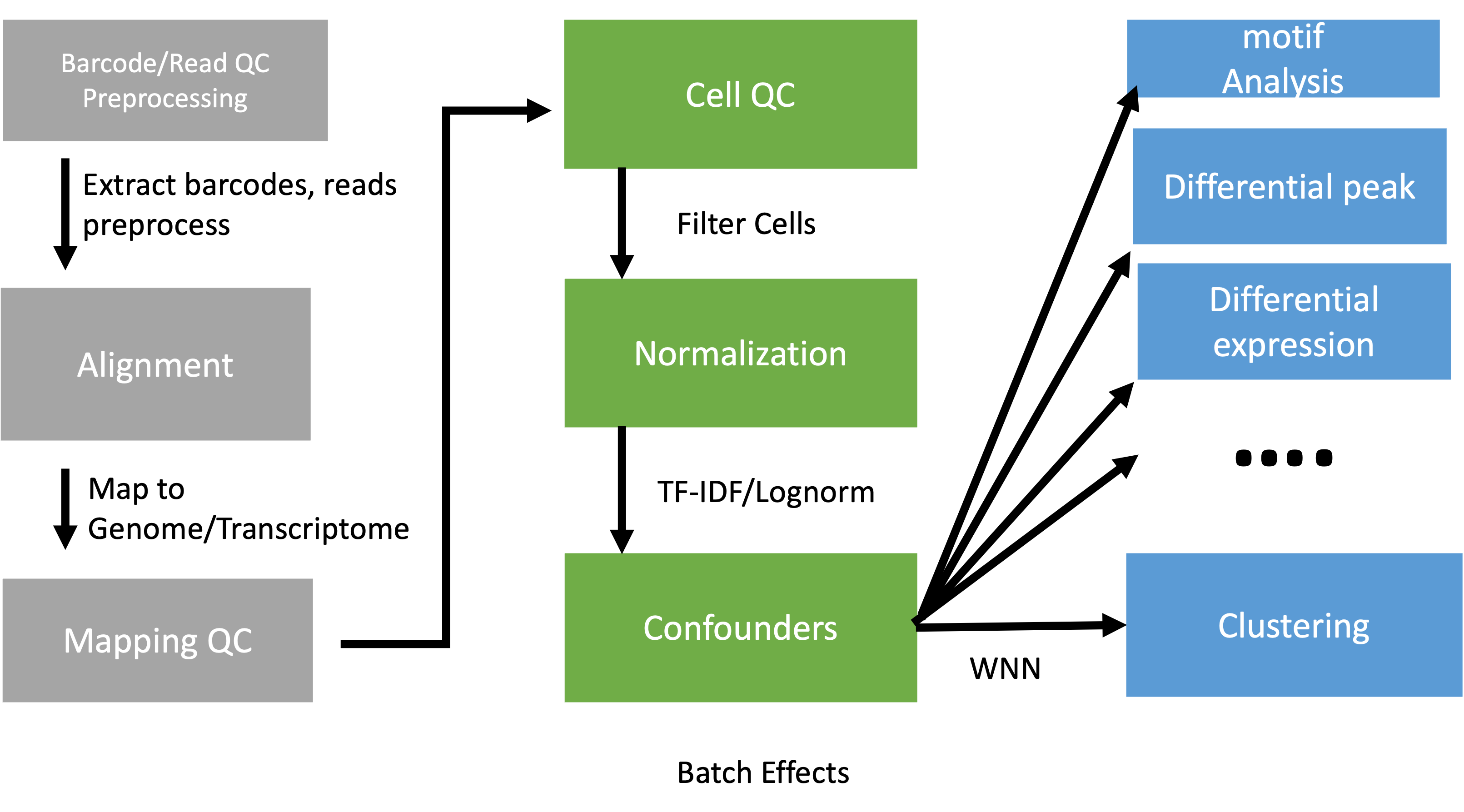
Data reduction
Log into tadpole and navigate to your directory on the /share/workshop space.
mkdir -p /share/workshop/scMultiome_workshop/$USER
cd /share/workshop/scMultiome_workshop/$USER
Request an interactive session from the scheduler so that we are not competing for resources on the head node.
srun -t 1-00:00:00 -c 4 -n 1 --mem 16000 --partition production --account workshop --reservation scworkshop --pty /bin/bash
Project set-up
Reads
mkdir -p /share/workshop/scMultiome_workshop/$USER/00-RawData
cd /share/workshop/scMultiome_workshop/$USER/00-RawData
ln -s /share/workshop/scMultiome_workshop/Data/fastqs/* .
Software
Before getting started, we need to make sure that we have the cellranger-arc software in our path. This can be done one of three ways:
- Module load:
module load cellranger-arc. This will only work on a cluster with modules for software management. - Add the location of a previously downloaded cellranger-arc build to our path:
export PATH=/share/workshop/scMultiome_workshop/software/cellranger-arc.2.0.2/bin:$PATH. This will not work if you don’t have a copy of cellranger somewhere on the system. - Download cellranger-arc to the current directory and add to our path:
wget -O cellranger-arc-2.0.2.tar.gz "https://cf.10xgenomics.com/releases/cell-arc/cellranger-arc-2.0.2.tar.gz?Expires=1718365082&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=kgwqjJ-xZv7YEXQVqCOgqSMe37sA40TspKfByqZ3raseybCLkm4NPWfA6pZWzSfKajzUdwI8lt67bH9TF2HGHF2qXLy5dniVehAiup-ZECQnArP~pjg-L607h8b4Id5cFZwSVH2ZN16JOlhGYl19v5yPZQZJbCDDQoiw62N~QdOcKkR-qeNrifU1sIH1k4GptBIDJDznu~dmKZs1RGaeJPOAUaFs1qWAVPJeRn2WaNUIuUnnAO6FrXWZr3gtqxtKMzY7f0qo5naBIelk3cjEmPNkTRvTlHgd940o-YVZi96lMzGISapxiCYIJ325nTTnWd7aEwLAPDuRfL38j4yTJA__"
tar -xzvf cellranger-arc-2.0.2.tar.gz
Reference
There are prebuilt human and mouse reference packages for use with Cell Ranger ARC, which we will be using in this workshop. For other species, or to create a custom reference, one can use cellranger-arc mkref.
cellranger-arc mkref
- Reference genome fasta (single species, primary assembly)
- Annotation gtf: should be filtered properly to remove low confidence features and to reduce the number of reads that map to overlapping regions of two genes
- Optional, transcription factor binding motif in position frequency matrices (JASPAR)
Config file to specify the custom references follows this example. The reference generated using cellranger-arc mkref can be used for cellranger, cellranger-atac, cellranger-arc.
The following code will generate a reference with cellranger-atac mkref. This takes a while, and is not used in this workshop. When using this code, please ensure that your FASTA and GTF files are appropriate versions, downloading up to date files as necessary.
cd /share/workshop/scMultiome_workshop/$USER
cellranger-arc mkref \
--config=path/to/config
Additional instructions for building ARC references can be found here.
Downloading prebuilt Cell Ranger reference
Since we are working with human data in this workshop, let’s download the prebuilt reference.
cd /share/workshop/scMultiome_workshop/$USER/
wget "https://cf.10xgenomics.com/supp/cell-arc/refdata-cellranger-arc-GRCh38-2020-A-2.0.0.tar.gz"
tar -xzvf refdata-cellranger-arc-GRCh38-2020-A-2.0.0.tar.gz
rm refdata-cellranger-arc-GRCh38-2020-A-2.0.0.tar.gz
Running cellranger-arc count
For experiments with multiome libraries, cellranger-arc count allows gene expression, and ATAC libraries from the same experiment to be processed simultaneously. Detailed descriptions of cellranger-arc count and cellranger-arc can be found on the 10x website.
Input
The call to cellranger-arc count requires a config file that provides the location of the fastq files for both the ATAC and gene expression libraries.
| Column | Description |
|---|---|
| fastqs | Directory that holds fastq files. Generally, this will be the fastq_path folder generated by cellranger mkfastq. |
| sample | The Illumina sample name. Generally, this will be as specified in the sample sheet supplied to mkfastq or bcl2fastq. |
| library_type | A description for the corresponding library type. |
cellranger-arc count \
--id=sampleID \
--libraries=${cwd}/config.csv \
--reference=refdata-cellranger-arc-GRCh38-2020-A-2.0.0
Output
There are a lot of files created in the output folder, including:
- web_summary.html - similar to gene expression
- summary.csv - run summary metrics, similar to gene expression
- filtered_feature_bc_matrix - gene expression and ATAC peak features
- gex_molecule_info.h5 - GEX Per molecule information
- atac_fragment.tsv.gz - atac fragment information
- atac_peaks.bed - atac peak locations
- atac_cut_sites.bigwig - ATAC smoothed transposition site track
- atac_peak_annotaion.tsv - ATAC peak annotations based on proximal genes
- BAMs - alignment files for ATAC and GEX
Running cellranger-arc aggr
cellranger-arc aggr provides one option of analyzing scATAC-Seq libraries from multiple datasets, with limited types of analyses.
library_id,atac_fragments,per_barcode_metrics,gex_molecule_info
sample1,sample1/outs/fragments.tsv.gz,sample1/outs/per_barcode_metrics.csv,sample1/outs/gex_molecule_info.h5
sample2,sample2/outs/fragments.tsv.gz,sample2/outs/per_barcode_metrics.csv,sample2/outs/gex_molecule_info.h5
cellranger-arc aggr \
--id=combined \
--csv=config.csv \
--reference=refdata-cellranger-arc-GRCh38-2020-A-2.0.0 \
--localcores=4 \
--normalize=none \
--dim-reduce=lsa \
--localmem=4
We are going to use the cellranger-arc results from another data for further analysis, and they are inside /share/workshop/scMultiome_workshop/cellranger_outs/.
On your local laptop/desktop, please create a project directory where you will do the rest of the analysis in. Please download the fragments.tsv.gz to your local laptop and keep the data structure. For example, having a directory structure like A001-C-007/outs/fragments.tsv.gz.
Download the analysis preparation Rmd file for the next section
wget https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2024-June-Single-Cell-RNA-Seq-Analysis/main/data_analysis/scMultiome_analysis_Part1.Rmd
Download the cellranger-arc results for downstream analysis
Please open another terminal on your laptop/desktop. Without logging into tadpole, create a folder for this workshop. Then go to this folder and download the cellranger outputs to it.
scp username@tadpole.genomecenter.ucdavis.edu:/share/workshop/scMultiome_workshop/Data/cellranger_outs.zip .
scp username@tadpole.genomecenter.ucdavis.edu:/share/workshop/scMultiome_workshop/Data/*.rds .