
Weighted Gene Correlation Network Analysis (WGCNA)
WGCNA analysis is an unsupervised machine learning technique. It is a powerful approach that captures the co-expression patterns from the correlation matrix. Rather than focusing on individual genes, such as in differential expression analysis we covered earlier in this workshop, it looks for clusters (modules) of genes that are connected based on the similarities in their expression patterns. These modules often correspond to biological pathways, disease states, cell types, tissue types,…
When applied to multiomics data, WGCNA can reduce each dataset down to a manageable number of features for a more interpretable analysis.
| Technique | Differential Expression | WGCNA |
|---|---|---|
| Approach | Compares gene expression between conditions | Identifies co-expressed gene modules |
| Base of analysis | Individual gene expression | Correlation of gene expression |
| Strengths | Straightforward interpretation with clear gene lists (ideally) | Systems-level view of gene relationships |
| Good for identifying candidate biomarkers | Dimensionality reduction which reduces multiple testing burden | |
| Can work with smaller sample sizes | Module eigengenes are robust and less noisy | |
| Easy to validate individual genes experimentally | Useful for discovering novel pathways | |
| Weaknesses | Treats genes independently, ignoring networks | Requires larger sample sizes (>15-20) |
| Arbitrary cutoffs may miss subtle but meaningful changes | Sensitive to parameter choices | |
| Doesn't reveal functional gene relationships | Correlation doesn't imply causation | |
| Can produce overwhelming gene lists, or empty lists | Modules may be hard to interpret biologically | |
| Sensitive to noise at individual gene level | Important non-co-expressing genes end up in 'grey' module | |
| Best for | Identifying specific genes that change between groups | Discovering gene network structure |
| Finding candidate biomarkers | Understanding coordinated biological processes | |
| Small sample sizes | Reduce data dimension |
Basic Steps of WGCNA
- Normalization, variance stabilization and filtering
- Determine soft-thresholding power for network construction
- Build gene co-expression network
- Explore gene modules
Install necessary R packages.
library(WGCNA)
library(ComplexHeatmap)
library(viridisLite)
library(matrixStats)
library(edgeR)
library(ggplot2)
library(dplyr)
library(tidyr)
library(clusterProfiler)
1. Read in expression table, normalization, variance stablization and filtering
We are going to use a different dataset for this exercise. The dataset has been used in 2 publications on study of cocaine’s effect on gene expression: 1 and 2. The experiment assigned male mice to one of six groups: saline or cocaine SA + 24 hr WD; or saline/cocaine SA + 30 d WD + an acute saline/cocaine challenge within the previous drug-paired context. RNASeq was conducted on six interconnected brain reward regions. We are going to use the data from 1 brain region, nucleus accumbens.
First, we are going to download the counts table and the metadata table for this dataset.
download.file("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2026-March-RNA-Seq-Analysis/master/datasets/NAc_counts.txt.gz", "NAc_counts.txt.gz")
download.file("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2026-March-RNA-Seq-Analysis/master/datasets/NAc_metadata.txt.gz", "NAc_metadata.txt.gz")
counts <- read.table("NAc_counts.txt.gz", header = T, check.names = F)
metadata <- read.table("NAc_metadata.txt.gz", header = T, sep = "\t", check.names = F)
rownames(metadata) <- metadata$sample_names
identical(colnames(counts), metadata$sample_names)
## [1] TRUE
Then we are going to use the same approach that we have learned earlier to normalize the data and perform variance stablization.
d0 <- DGEList(counts)
d0 <- calcNormFactors(d0)
expr0 <- cpm(d0, log = T)
Next we will filter out low expressed genes, using the filterByExpr function in edgeR.
keep <- filterByExpr(d0, model.matrix(~group, data = metadata))
expr0 <- expr0[keep,]
Unlike many other programs, WGCNA works on a matrix where the rows are samples and the columns are genes.
expr <- t(expr0)
2. Select soft-thresholding power
The soft-thresholding power parameter is crucial for network construction. The goal of a good soft-thresholding power is to achive scale-free topology while maintaining reasonable connectivity within the network. Higher powers lead to higher suppression of weak correlations and create sparser networks with fewer but stronger connections. The optimal soft-thresholding power is dataset dependent because it reflects the underlying correlation structure of the data.
In a scale-free network, most nodes (genes) have few connections and a few nodes have lots of connections.
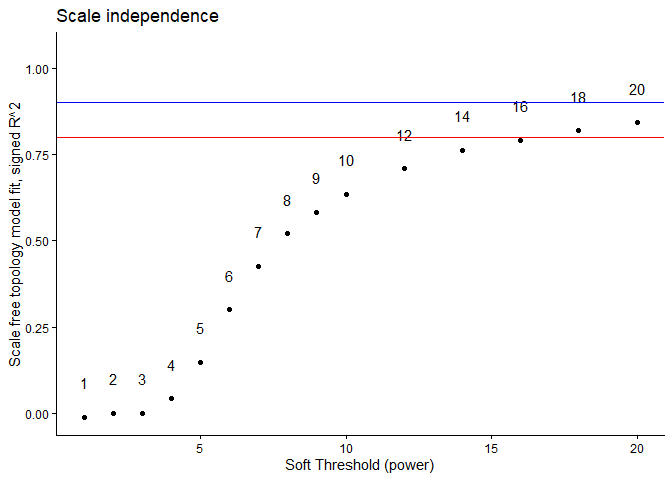
The recommended selection criteria is to achieve $R^2$ $g \ge f$ 0.8-0.9, as well as a reasonable mean connectivity (< 200-300). The function pickSoftThreshold scans through a series of soft-thresholding powers and produces the corresponding network characteristics that can be used for the selection.
We will use Tukey’s robust biweight correlation to estimate correlations.
sft <- pickSoftThreshold(expr, dataIsExpr = T, networkType = "signed", corFnc = "bicor", RsquaredCut = 0.8)
## Warning: executing %dopar% sequentially: no parallel backend registered
## Power SFT.R.sq slope truncated.R.sq mean.k. median.k. max.k.
## 1 1 1.06e-02 3.56000 0.942 8560.0 8560.0 9130
## 2 2 3.52e-07 -0.00785 0.910 4650.0 4640.0 5340
## 3 3 4.40e-04 -0.15600 0.868 2680.0 2660.0 3410
## 4 4 4.31e-02 -0.99300 0.862 1630.0 1610.0 2360
## 5 5 1.49e-01 -1.27000 0.894 1040.0 1010.0 1710
## 6 6 3.00e-01 -1.55000 0.914 685.0 661.0 1310
## 7 7 4.27e-01 -1.75000 0.924 468.0 443.0 1040
## 8 8 5.20e-01 -1.85000 0.934 329.0 304.0 850
## 9 9 5.83e-01 -1.92000 0.938 237.0 213.0 706
## 10 10 6.35e-01 -1.97000 0.948 175.0 152.0 596
## 11 12 7.09e-01 -2.01000 0.965 101.0 81.9 441
## 12 14 7.63e-01 -2.04000 0.976 62.4 46.0 337
## 13 16 7.92e-01 -2.05000 0.981 40.4 27.1 265
## 14 18 8.19e-01 -2.05000 0.987 27.3 16.4 212
## 15 20 8.43e-01 -2.03000 0.990 19.0 10.3 172
Let’s plot $R^2$.
metrics <- data.frame(sft$fitIndices) %>% dplyr::mutate(model_fit = -sign(slope) * SFT.R.sq)
metrics
## Power SFT.R.sq slope truncated.R.sq mean.k. median.k.
## 1 1 1.062245e-02 3.564097269 0.9423160 8560.41223 8559.67003
## 2 2 3.521658e-07 -0.007847019 0.9098681 4650.34812 4638.36624
## 3 3 4.395832e-04 -0.155568601 0.8684206 2684.76028 2663.77189
## 4 4 4.310165e-02 -0.993358290 0.8617788 1631.87839 1608.28226
## 5 5 1.491049e-01 -1.274202968 0.8941794 1036.83887 1012.93505
## 6 6 3.000956e-01 -1.554853819 0.9135453 684.62787 660.57937
## 7 7 4.267064e-01 -1.748495279 0.9243824 467.53029 443.35711
## 8 8 5.201339e-01 -1.852753924 0.9337967 328.83526 304.46330
## 9 9 5.829592e-01 -1.920326235 0.9383803 237.36129 213.35742
## 10 10 6.348166e-01 -1.969297733 0.9480853 175.29177 152.40036
## 11 12 7.092466e-01 -2.006449010 0.9652948 101.32673 81.93968
## 12 14 7.625351e-01 -2.035140119 0.9762534 62.41645 46.02759
## 13 16 7.918794e-01 -2.054283628 0.9806997 40.44048 27.05765
## 14 18 8.192845e-01 -2.046627547 0.9868942 27.29113 16.42197
## 15 20 8.425328e-01 -2.032690202 0.9903099 19.04160 10.27170
## max.k. model_fit
## 1 9126.9845 -1.062245e-02
## 2 5337.4796 3.521658e-07
## 3 3413.1037 4.395832e-04
## 4 2356.7899 4.310165e-02
## 5 1710.1271 1.491049e-01
## 6 1308.6426 3.000956e-01
## 7 1041.6463 4.267064e-01
## 8 849.5948 5.201339e-01
## 9 706.4062 5.829592e-01
## 10 596.4715 6.348166e-01
## 11 440.6175 7.092466e-01
## 12 337.1290 7.625351e-01
## 13 264.6360 7.918794e-01
## 14 211.8230 8.192845e-01
## 15 172.1854 8.425328e-01
ggplot(metrics, aes(x = Power, y = model_fit, label = Power)) +
geom_point() + geom_text(nudge_y = 0.1) +
geom_hline(yintercept = 0.8, col = "red") +
geom_hline(yintercept = 0.9, col = "blue") +
ylim(c(min(metrics$model_fit), 1.05)) + xlab("Soft Threshold (power)") +
ylab("Scale free topology model fit, signed R^2") +
ggtitle("Scale independence") + theme_classic()
ggplot(metrics, aes(x = Power, y = mean.k., label = Power)) +
geom_point() + geom_text(nudge_y = 0.1) +
geom_hline(yintercept = 200, col = "red") +
xlab("Soft Threshold (power)") +
ylab("Mean connectivity") +
ggtitle("Mean connectivity") + theme_classic()
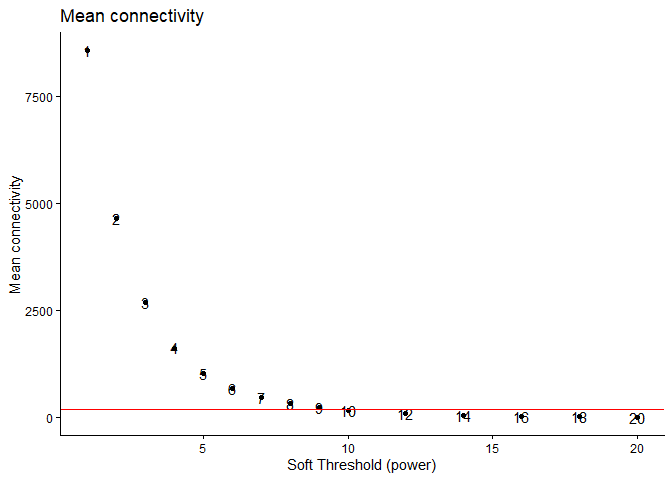
Based on the above plot, we are going to pick the soft-thresholding power to be 12. With a power of 12, the R^2 value is close to 0.8 while still maintaining a reasonable amount of connectivity.
3. Build gene co-expression network
nwk <- blockwiseModules(expr, power = 12, minModuleSize = 30, TOMType = "signed", corType = "bicor", mergeCutHeight = 0.4)
4. Explore the modules detected
First, let’s visualize the gene network using a dendrogram
plotDendroAndColors(nwk$dendrograms[[1]], nwk$colors[nwk$blockGenes[[1]]], "Module Colors",
dendroLabels = F, addGuide = T, main = "Gene dendrogram and module colors")
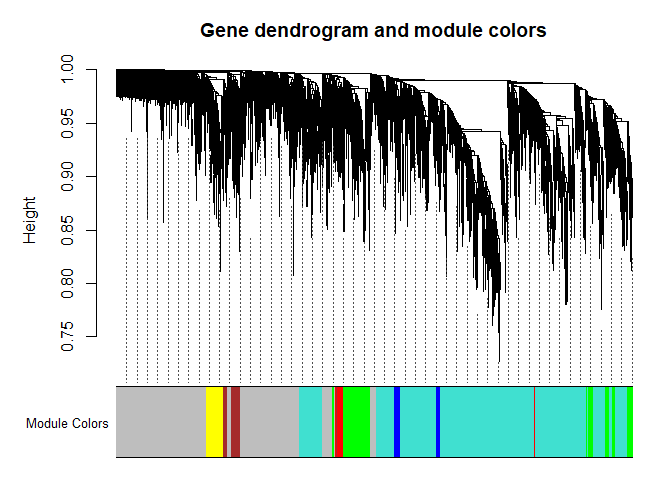
Let’s look at what genes are in each module and merge in annotation information
module.members <- data.frame(Gene.stable.ID = names(nwk$colors), Module = nwk$colors)
anno <- read.delim("ensembl_mm_115.txt")
module.members <- left_join(module.members, anno, by = "Gene.stable.ID")
module.members <- arrange(module.members, Module)
head(module.members)
## Gene.stable.ID Module Gene.stable.ID.version
## 1 ENSMUSG00000000296 black ENSMUSG00000000296.9
## 2 ENSMUSG00000000552 black ENSMUSG00000000552.11
## 3 ENSMUSG00000000753 black ENSMUSG00000000753.16
## 4 ENSMUSG00000000792 black ENSMUSG00000000792.3
## 5 ENSMUSG00000001507 black ENSMUSG00000001507.17
## 6 ENSMUSG00000001870 black ENSMUSG00000001870.17
## Gene.description
## 1 tumor protein D52-like 1 [Source:MGI Symbol;Acc:MGI:1298386]
## 2 zinc finger protein 385A [Source:MGI Symbol;Acc:MGI:1352495]
## 3 serine (or cysteine) peptidase inhibitor, clade F, member 1 [Source:MGI Symbol;Acc:MGI:108080]
## 4 solute carrier family 5 (sodium iodide symporter), member 5 [Source:MGI Symbol;Acc:MGI:2149330]
## 5 integrin alpha 3 [Source:MGI Symbol;Acc:MGI:96602]
## 6 latent transforming growth factor beta binding protein 1 [Source:MGI Symbol;Acc:MGI:109151]
## Chromosome.scaffold.name Gene.start..bp. Gene.end..bp. Gene.name
## 1 10 31208372 31321954 Tpd52l1
## 2 15 103222322 103248520 Zfp385a
## 3 11 75300595 75313527 Serpinf1
## 4 8 71335533 71345401 Slc5a5
## 5 11 94935300 94967627 Itga3
## 6 17 75312563 75699507 Ltbp1
## Gene...GC.content Gene.type
## 1 40.19 protein_coding
## 2 50.68 protein_coding
## 3 49.44 protein_coding
## 4 54.85 protein_coding
## 5 50.82 protein_coding
## 6 42.66 protein_coding
write.table(module.members, file = "module_memberships.txt", sep = "\t", quote = FALSE, row.names = FALSE)
The function chooseTopHubInEachModule can be used to identify highly connected “hub” genes:
hubs <- chooseTopHubInEachModule(expr, colorh = nwk$colors)
# Make into a data frame and merge in annotation
hub.data <- data.frame(Module = names(hubs), TopHubGene = hubs) %>%
left_join(anno, by = c("TopHubGene" = "Gene.stable.ID")) %>%
select(Module, TopHubGene, Gene.name, Gene.description)
hub.data
## Module TopHubGene Gene.name
## 1 black ENSMUSG00000030268 Bcat1
## 2 blue ENSMUSG00000022452 Smdt1
## 3 brown ENSMUSG00000038170 Pde4dip
## 4 green ENSMUSG00000034156 Tspoap1
## 5 greenyellow ENSMUSG00000036634 Mag
## 6 magenta ENSMUSG00000043164 Tmem212
## 7 pink ENSMUSG00000062393 Dgkk
## 8 purple ENSMUSG00000026824 Kcnj3
## 9 red ENSMUSG00000029054 Gabrd
## 10 turquoise ENSMUSG00000027104 Atf2
## 11 yellow ENSMUSG00000030062 Rpn1
## Gene.description
## 1 branched chain aminotransferase 1, cytosolic [Source:MGI Symbol;Acc:MGI:104861]
## 2 single-pass membrane protein with aspartate rich tail 1 [Source:MGI Symbol;Acc:MGI:1916279]
## 3 phosphodiesterase 4D interacting protein (myomegalin) [Source:MGI Symbol;Acc:MGI:1891434]
## 4 TSPO associated protein 1 [Source:MGI Symbol;Acc:MGI:2450877]
## 5 myelin-associated glycoprotein [Source:MGI Symbol;Acc:MGI:96912]
## 6 transmembrane protein 212 [Source:MGI Symbol;Acc:MGI:2685410]
## 7 diacylglycerol kinase kappa [Source:MGI Symbol;Acc:MGI:3580254]
## 8 potassium inwardly-rectifying channel, subfamily J, member 3 [Source:MGI Symbol;Acc:MGI:104742]
## 9 gamma-aminobutyric acid (GABA) A receptor, subunit delta [Source:MGI Symbol;Acc:MGI:95622]
## 10 activating transcription factor 2 [Source:MGI Symbol;Acc:MGI:109349]
## 11 ribophorin I [Source:MGI Symbol;Acc:MGI:98084]
Gene ontology enrichment can be used to characterize genes in a module. We use clusterProfiler here but topGO can also be used.
module.use <- "purple"
module.genes <- names(nwk$colors)[nwk$colors == module.use]
module.genes.entrez <- bitr(module.genes, fromType = "ENSEMBL", toType = "ENTREZID", OrgDb = "org.Mm.eg.db")
##
## 'select()' returned 1:many mapping between keys and columns
go <- enrichGO(module.genes.entrez$ENTREZID, OrgDb = "org.Mm.eg.db")
go <- setReadable(go, OrgDb = "org.Mm.eg.db")
as.data.frame(go)
## ID Description GeneRatio BgRatio
## GO:0005179 GO:0005179 hormone activity 6/137 146/23909
## GO:0051428 GO:0051428 peptide hormone receptor binding 3/137 21/23909
## GO:0000149 GO:0000149 SNARE binding 5/137 118/23909
## GO:0005184 GO:0005184 neuropeptide hormone activity 3/137 29/23909
## GO:0045499 GO:0045499 chemorepellent activity 3/137 29/23909
## GO:0051427 GO:0051427 hormone receptor binding 3/137 30/23909
## GO:0160041 GO:0160041 neuropeptide activity 3/137 30/23909
## GO:0019905 GO:0019905 syntaxin binding 4/137 71/23909
## RichFactor FoldEnrichment zScore pvalue p.adjust
## GO:0005179 0.04109589 7.171983 5.678708 0.0001993704 0.03397314
## GO:0051428 0.14285714 24.931178 8.328809 0.0002269615 0.03397314
## GO:0000149 0.04237288 7.394841 5.286442 0.0005981516 0.03397314
## GO:0005184 0.10344828 18.053612 6.975852 0.0006029920 0.03397314
## GO:0045499 0.10344828 18.053612 6.975852 0.0006029920 0.03397314
## GO:0051427 0.10000000 17.451825 6.844878 0.0006671929 0.03397314
## GO:0160041 0.10000000 17.451825 6.844878 0.0006671929 0.03397314
## GO:0019905 0.05633803 9.832014 5.657871 0.0007446167 0.03397314
## qvalue geneID Count
## GO:0005179 0.02978467 Fndc5/Vip/Igf1/Adcyap1/Cck/Ccn3 6
## GO:0051428 0.02978467 Vip/Adcyap1/Cck 3
## GO:0000149 0.02978467 Napb/Snap91/Cplx1/Syt1/Cplx3 5
## GO:0005184 0.02978467 Vip/Adcyap1/Cck 3
## GO:0045499 0.02978467 Sema3a/Sema7a/Nrg3 3
## GO:0051427 0.02978467 Vip/Adcyap1/Cck 3
## GO:0160041 0.02978467 Vip/Adcyap1/Cck 3
## GO:0019905 0.02978467 Napb/Cplx1/Syt1/Cplx3 4
Let’s take a look at the correlations among the modules by using the module eigengenes. We use a Spearman (nonparametric) correlation.
All WGCNA module names are valid R color names that can be used in plotting.
MEs <- orderMEs(nwk$MEs)
cor_MEs <- cor(MEs, method = "spearman")
colors <- gsub("ME", "", rownames(cor_MEs))
names(colors) <- rownames(cor_MEs)
ra <- rowAnnotation(Module = rownames(cor_MEs), col = list(Module = colors), show_legend = FALSE)
ca <- columnAnnotation(Module = rownames(cor_MEs), col = list(Module = colors), show_legend = FALSE)
ht <- Heatmap(cor_MEs, name = "Spearman\nCorrelation", column_title = "Module Eigengene Correlation", left_annotation = ra, top_annotation = ca)
draw(ht)
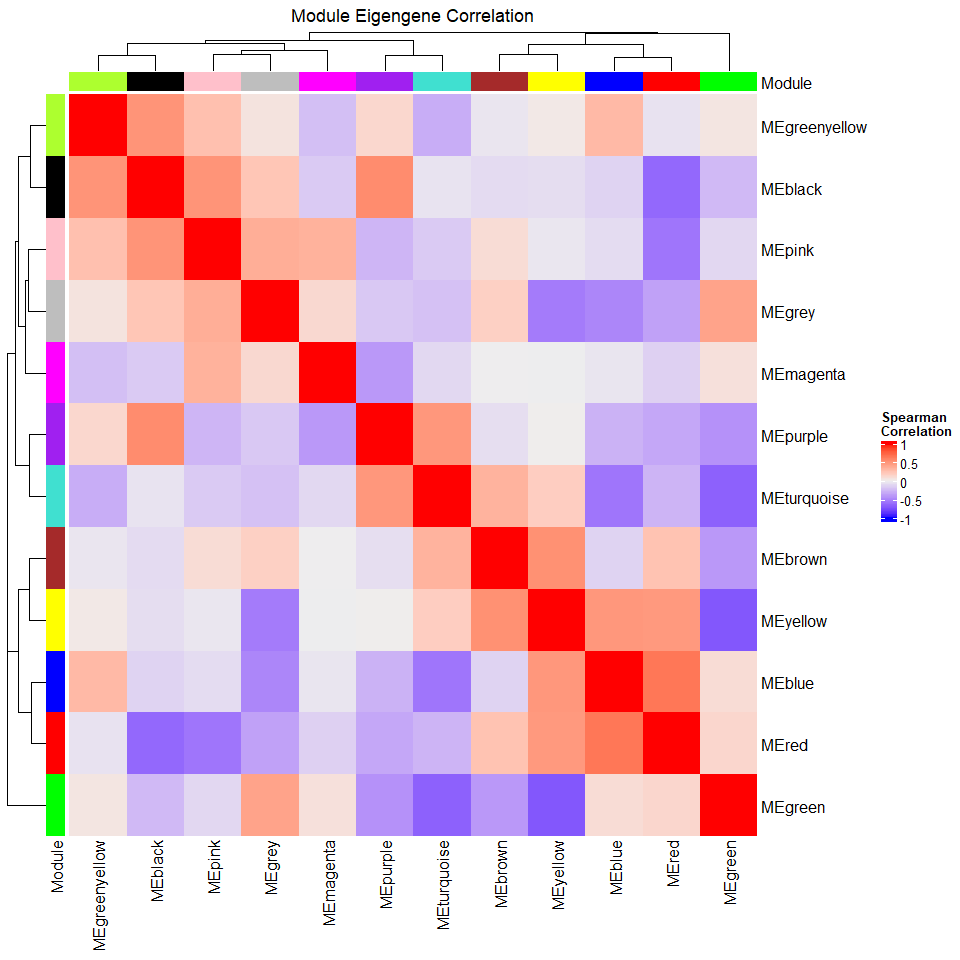
For the above plot, bear in mind that a simple correlation coefficient may not be appropriate if your data have repeated measures or other complications. Any analysis on the eigengenes requires the same thoughtfulness as an analysis on your original data.
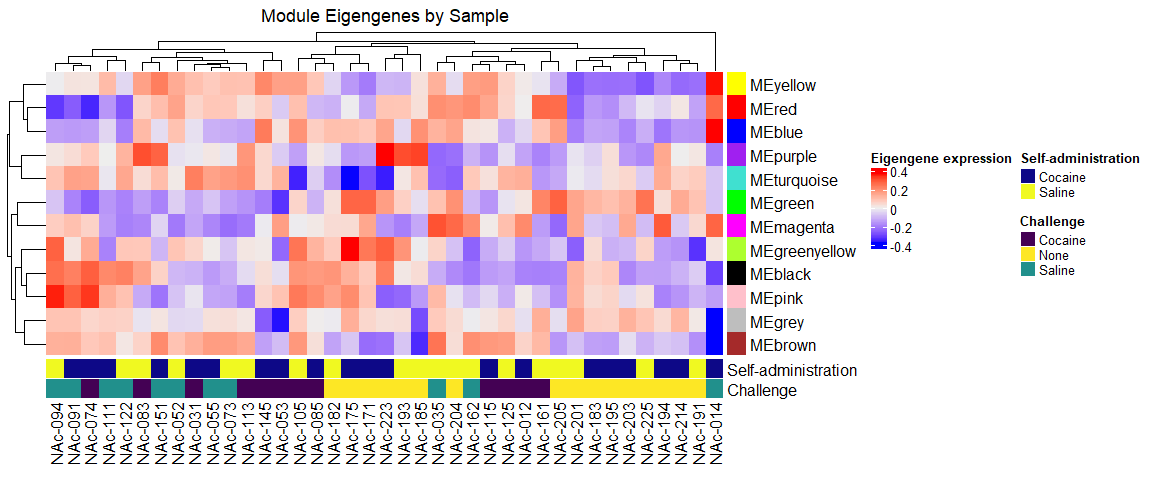
Let’s take a look at samples at this reduced dimension using the module eigengenes. We can plot a heatmap of the eigengenes by samples exactly as we would plot genes by samples for the original data.
admins <- unique(metadata$`self-administration`)
admin_colors <- setNames(plasma(length(admins)), admins)
challenges <- unique(metadata$challenge)
challenge_colors <- setNames(viridis(length(challenges)), challenges)
ann_colors <- list(`Self-administration` = admin_colors,
Challenge = challenge_colors)
row_colors <- gsub("ME", "", colnames(MEs))
names(row_colors) <- colnames(MEs)
ca <- columnAnnotation(`Self-administration` = metadata$`self-administration`, Challenge = metadata$challenge, col = list(`Self-administration` = admin_colors,
Challenge = challenge_colors))
ra <- rowAnnotation(Module = colnames(MEs), col = list(Module = row_colors), show_legend = FALSE, show_annotation_name = FALSE)
ht <- Heatmap(t(MEs), bottom_annotation = ca, right_annotation = ra, name = "Eigengene expression", column_title = "Module Eigengenes by Sample")
draw(ht)
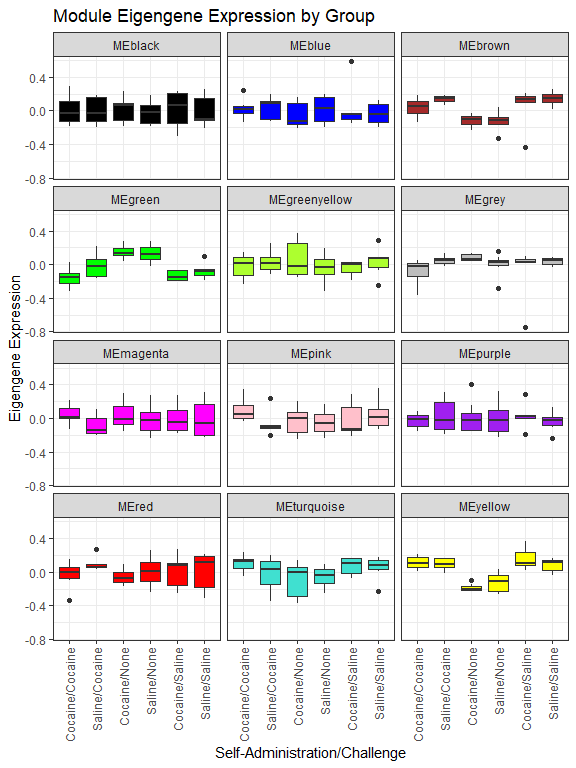
We can make boxplots of module eigengenes by experimental group:
# Turn MEs into long dataset for plotting
tmp <- MEs
tmp$sample_names <- rownames(tmp)
longMEs <- pivot_longer(tmp, cols = -sample_names, names_to = "Module", values_to = "Expression") %>%
# Merge in metadata
left_join(metadata, by = "sample_names")
longMEs$group2 <- interaction(longMEs$`self-administration`, longMEs$challenge, sep = "/")
ggplot(longMEs, aes(x = group2, y = Expression, fill = Module)) +
geom_boxplot() +
facet_wrap(~Module, nrow = 4) +
scale_fill_manual(breaks = colnames(MEs), values = gsub("ME", "", colnames(MEs))) + labs(x = "Self-Administration/Challenge", y = "Eigengene Expression", title = "Module Eigengene Expression by Group") +
theme_bw() +
theme(legend.position = "none", axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1))
Multiomics analysis
For multiomics analyses, WGCNA can be useful for reducing each dataset to a few features before combining them. If, for example, you had transcriptomics and metabolomics data, you might do the following:
-
Perform WGCNA on your transcriptomics data as shown in this document
-
Separately, perform WGCNA on your metabolomics data as shown in this document EXCEPT doing normalization and filtering appropriate for metabolomics data.
-
Use the module eigengenes (eigenmetabolites?) from the transcriptomics data and metabolomics data in an integrated analysis. For example, you could look at the correlation between the eigengenes from each dataset.