
Raw Data Preprocessing
This document assumes project_setup has been completed.
cd /share/workshop/gwas_workshop/$USER/gwas_example
Why Preprocess Reads
We have found that aggressively “cleaning” and preprocessing of reads can make a large difference to the speed and quality of mapping and assembly results. Preprocessing your reads means:
- Removing bases of unwanted sequence (Ex. vectors, adapter, primer sequence, polyA tails).
- Remove reads that are not of primary interest (contamination).
- Remove low quality bases or N characters.
- Remove reads originating from PCR duplication.
- Remove too short reads.
Preprocessing also produces a number of statistics about the samples. These can be used to evaluate sample-to-sample consistency.
Many read preprocessing strategies over time
- Identity and remove contaminant and vector reads
- Reads which appear to fully come from extraneous sequence should be removed.
- Quality trim/cut
- “End” trim a read until the average quality > Q (Lucy)
- Remove any read with average quality < Q
- Eliminate singletons/duplicates
- If you have excess depth of coverage, and particularly if you have at least x-fold coverage where x is the read length, then eliminating singletons is a nice way of dramatically reducing the number of error-prone reads.
- Read which appear the same (particularly paired-end) are often more likely PCR duplicates and therefor redundant reads.
- Eliminate all reads (pairs) containing an “N” character
- If you can afford the loss of coverage, you might throw away all reads containing Ns.
- Identity and trim off adapter and barcodes if present
- Believe it or not, the software provided by Illumina, either does not look for, or does a mediocre job of, identifying adapters and removing them.
Many technical things happen between original sample and data. Preprocessing is working backwards through that process to get as close as we can to original sample.
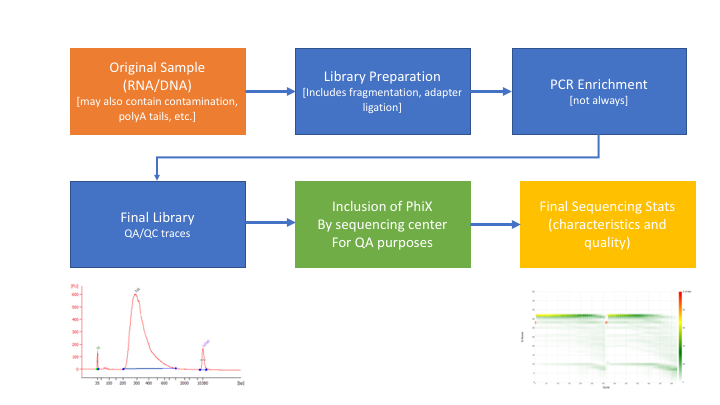
In order to better understand and preprocess our data set (and to determine the types of problems we might encounter), it is a good idea to learn what type of library prep kit was used, and how it works.
- Remove contaminants (at least PhiX).
- Remove PCR duplicates.
- Remove adapter sequences.
- Trim sequences (5’ and 3’) by quality score (I like Q20)
- Cleanup
- Remove any reads that are less then the minimum length parameter
- Produce preprocessing statistics
HTStream Streamed Preprocessing of Sequence Data
HTStream is a suite of preprocessing applications for high throughput sequencing data (ex. Illumina). A fast C++ implementation, designed with discreet functionality that can be pipelined together using standard Unix piping.
Benefits Include:
- No intermediate files, reducing storage footprint.
- Reduced I/O, files are only read in and written out once to disk.
- Handles both single end and paired end reads at the same time.
- Applications process reads at the same time allowing for process parallelization.
- Built on top of mature C++ Boost libraries to reduce bugs and memory leaks.
- Designed following the philosophy of Program Design in the UNIX Environment.
- Works with native Unix/Linux applications such as grep/sed/awk etc.
- Can build a custom preprocessing pipeline to fit the specific expectation of the data.
- A single JSON output per sample detailing the preprocessing statistics from each application.
HTStream achieves these benefits by using a tab delimited intermediate format that allows for streaming from application to application. This streaming creates some awesome efficiencies when preprocessing HTS data and makes it fully interoperable with other standard Linux tools.
A traditional preprocessing pipeline:
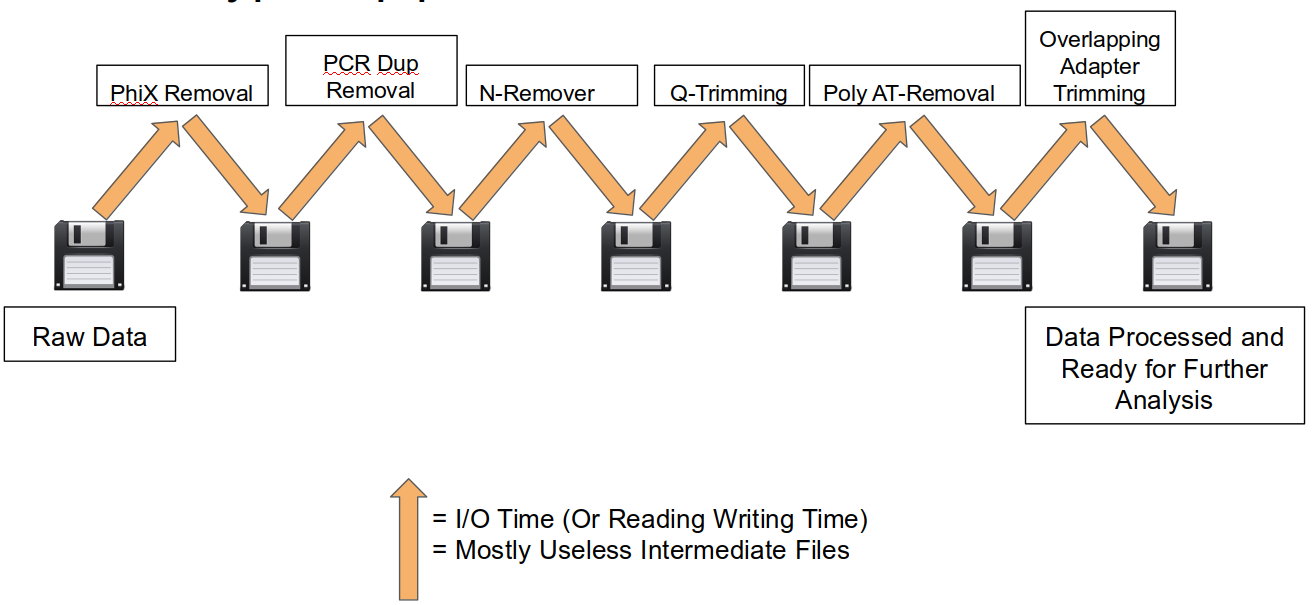
An HTStream preprocessing pipline:
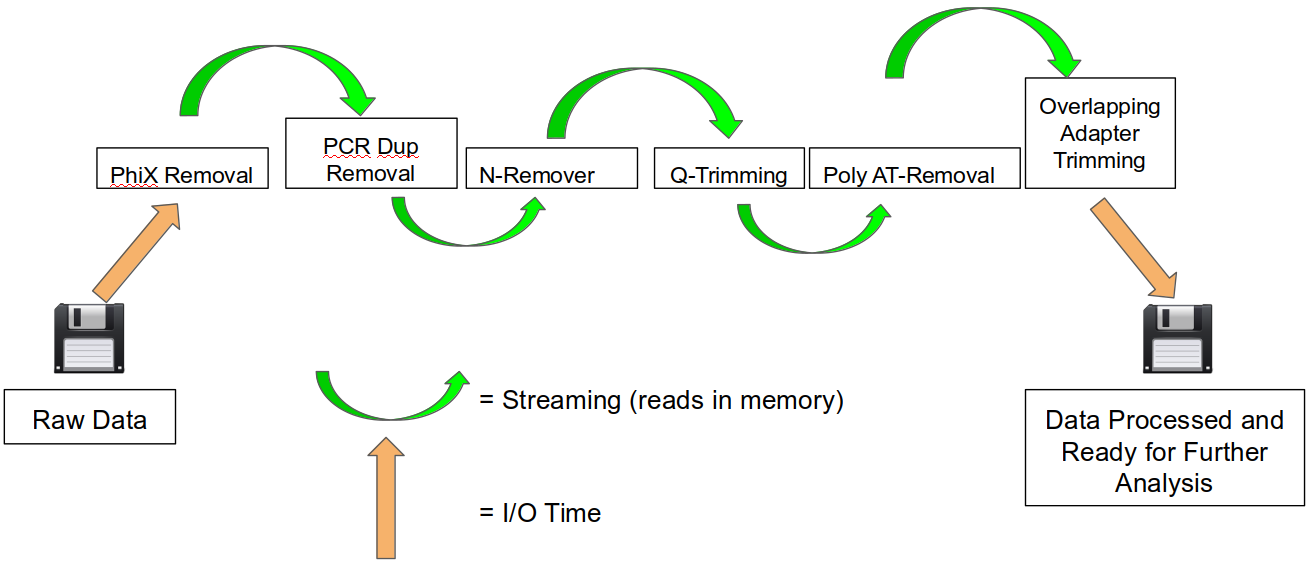
This approach also uses significantly less storage as there are no intermediate files. HTStream can do this by streaming a tab-delimited format called tab6.
Single end reads are 3 columns:
read1id read1seq read1qual
Paired end reads are 6 columns:
read1id read1seq read1qual read2id read2seq read2qual
HTStream applications
HTStream includes the following applications:
hts_AdapterTrimmer: Identify and remove adapter sequences.
hts_CutTrim: Discreet 5’ and/or 3’ basepair trimming.
hts_LengthFilter: Remove reads outside of min and/or max length.
hts_NTrimmer: Extract the longest subsequence with no Ns.
hts_Overlapper: Overlap paired end reads, removing adapters when present.
hts_PolyATTrim: Identify and remove polyA/T sequence.
hts_Primers: Identify and optionally remove 5’ and/or 3’ primer sequence.
hts_QWindowTrim: 5’ and/or 3’ quality score base trimming using windows.
hts_SeqScreener: Identify and remove/keep/count contaminants (default phiX).
hts_Stats: Compute read stats.
hts_SuperDeduper: Identify and remove PCR duplicates.
The source code and pre-compiled binaries for Linux can be downloaded and installed from the GitHub repository.
HTStream is also available on Bioconda, and there is even an image on Docker Hub.
HTStream was designed to be extensible. We continue to add new preprocessing routines and welcome contributions from collaborators.
If you encounter any bugs or have suggestions for improvement, please post them to issues.
HTStream tutorial
Start Group Exercise 1:
Running HTStream
Let’s run the first step of our HTStream preprocessing pipeline, which is always to gather basic stats on the read files. For now, we’re only going to run one sample through the pipeline.
When building a new pipeline, it is almost always a good idea to use a small subset of the data in order to speed up development. A small sample of reads will take seconds to process and help you identify problems that may have only been apparent after hours of waiting for the full data set to process.
-
Let’s start by first taking a small subsample of reads, so that our trial run through the pipeline goes really quickly.
cd /share/workshop/gwas_workshop/$USER/gwas_example mkdir HTS_testing cd HTS_testingThen create a small dataset.
zcat ../00-RawData/SL378587_S1_L001_R1_001.fastq.gz | head -4000 | gzip > SL378587_subset_R1.fastq.gz zcat ../00-RawData/SL378587_S1_L001_R2_001.fastq.gz | head -4000 | gzip > SL378587_subset_R2.fastq.gz ls -lSo we
zcat(uncompress and send to stdout), pipe|tohead(param -4000) then pipe togzipto recompress and name our files subset.- How many reads are we going to analyze in our subset?
-
Now we’ll run our first preprocessing step
hts_Stats, first loading the module and then looking at help.cd /share/workshop/gwas_workshop/$USER/gwas_example/HTS_testing module load htstream hts_Stats --helpjli@barbera:/share/workshop/gwas_workshop/jli/gwas_example$ hts_Stats --help HTStream <https://github.com/s4hts/HTStream> application: hts_Stats Version: v1.3.3 The hts_Stats app produce basic statistics about the reads in a dataset. Including the basepair composition and number of bases Q30. Standard Options: -v [ --version ] Version print -h [ --help ] Prints help documentation -N [ --notes ] arg Notes for the stats JSON -L [ --stats-file ] arg (=stats.json) Write to stats file name -A [ --append-stats-file ] arg Append to stats file name Input Options [default: tab6 format on stdin]: -1 [ --read1-input ] arg Read 1 paired end fastq input-2 [ --read2-input ] arg Read 2 paired end fastq input -U [ --singleend-input ] arg Single end read fastq input -I [ --interleaved-input ] arg Interleaved fastq input -T [ --tab-input ] arg Tab-delimited (tab6) input hts_Stats Output Options [default: tab6 format to stdout]: -F [ --force ] Forces overwrite of files -u [ --uncompressed ] Output uncompressed (not gzipped) files -f [ --fastq-output ] arg Output to Fastq files <PE AND/OR SE files> -i [ --interleaved-output ] arg Output to interleaved fastq files <INTERLEAVED PE AND/OR SE files> -t [ --tab-output ] arg Output to tab-delimited (tab6) file -z [ --unmapped-output ] arg Output to unmapped sam file Application Specific Options: </div>
- What version of hts_Stats is loaded?
-
Now lets run
hts_Statsand look at the output.hts_Stats -1 SL378587_subset_R1.fastq.gz \ -2 SL378587_subset_R2.fastq.gz \ -L SL378587.stats.json -f SL378587.stats- What parameters did we use? What do they do?
ls -lahtotal 400K drwxrwsr-x 2 jli workshop 7 Jul 12 15:19 . drwxrwsr-x 9 jli workshop 11 Jul 12 15:16 .. -rw-rw-r-- 1 jli workshop 53K Jul 12 15:19 SL378587.stats.json -rw-rw-r-- 1 jli workshop 68K Jul 12 15:19 SL378587.stats_R1.fastq.gz -rw-rw-r-- 1 jli workshop 73K Jul 12 15:19 SL378587.stats_R2.fastq.gz -rw-rw-r-- 1 jli workshop 68K Jul 12 15:17 SL378587_subset_R1.fastq.gz -rw-rw-r-- 1 jli workshop 73K Jul 12 15:17 SL378587_subset_R2.fastq.gz- Which files were generated from hts_Stats?
- Did stats change any of the data (are the contents of SL378587.stats_R1.fastq.gz identical to SL378587_subset_R1.fastq.gz)?
-
Lets look at the file SL378587.stats.json
less -S SL378587.stats.jsonThe logs generated by htstream are in JSON format, like a database format but meant to be readable.
-
Run
hts_SeqScreenerto remove PhiX contamination.hts_SeqScreener -1 SL378587_subset_R1.fastq.gz \ -2 SL378587_subset_R2.fastq.gz \ -L SL378587.SeqScreener.json -f SL378587.SeqScreener-
Which files were generated from hts_SeqScreener?
-
Take look at the file SL378587.SeqScreener.json
-
How many reads were identified as PhiX contamination?
-
What fraction of reads were identified as PhiX, do you think cleanup worked well for this sample?
-
Getting more advanced: Streaming multiple applications together
-
Lets try it out. First run hts_Stats and then hts_SeqScreener in a streamed fashion.
cd /share/workshop/gwas_workshop/$USER/gwas_example/HTS_testing hts_Stats -1 SL378587_subset_R1.fastq.gz \ -2 SL378587_subset_R2.fastq.gz \ -L SL378587.streamed.json | hts_SeqScreener -A SL378587.streamed.json \ -f SL378587.streamedNote the pipe,
|, between the two applications!Questions
-
What new parameters did we use here?
-
What parameter is SeqScreener using that specifies how reads are input?
-
Look at the file SL378587.streamed.json
-
Can you find the section for each program?
-
Were the programs run in the order you expected?
-
-
*hts_SeqScreener will screen out PhiX reads by default.
-
Stop Group Exercise 1
A DNA preprocessing pipeline
- hts_Stats: get stats on input raw reads
- hts_SeqScreener: screen out (remove) phiX
- hts_SuperDeduper: identify and remove PCR duplicates
- hts_AdapterTrimmer: identify and remove adapter sequence
- hts_QWindowTrim: remove poor quality bases
- hts_NTrimmer: trim to remove any remaining N characters
- hts_LengthFilter: use to remove all reads < 50bp
- hts_Stats: get stats on output cleaned reads
Why screen for phiX?
PhiX Control v3 is a common control in Illumina runs, and facilities may not tell you if/when PhiX has been spiked in. Since it does not have a barcode, in theory should not be in your data.
However:
- When we know PhiX has been spiked in, we find sequence every time.
- [update] When dual matched barcodes are used, then almost zero phiX reads can be identified.
- When we know that PhiX has not been spiked in, we rarely find matching sequence.
For variant analysis (any mapping based technique) it is not critical to remove, but for sequence assembly it is (and will often assemble into a full-length PhiX genome). Unless you are sequencing PhiX, it is noise, so its better safe than sorry to screen for it every time.
Removing PCR duplicates with hts_SuperDeduper.
Removing duplicates is standard practice in variant analysis. It tells you alot about the original complexity of each sample and potential impact of sequencing depth.
However, I would never do PCR duplicate removal on single-end reads
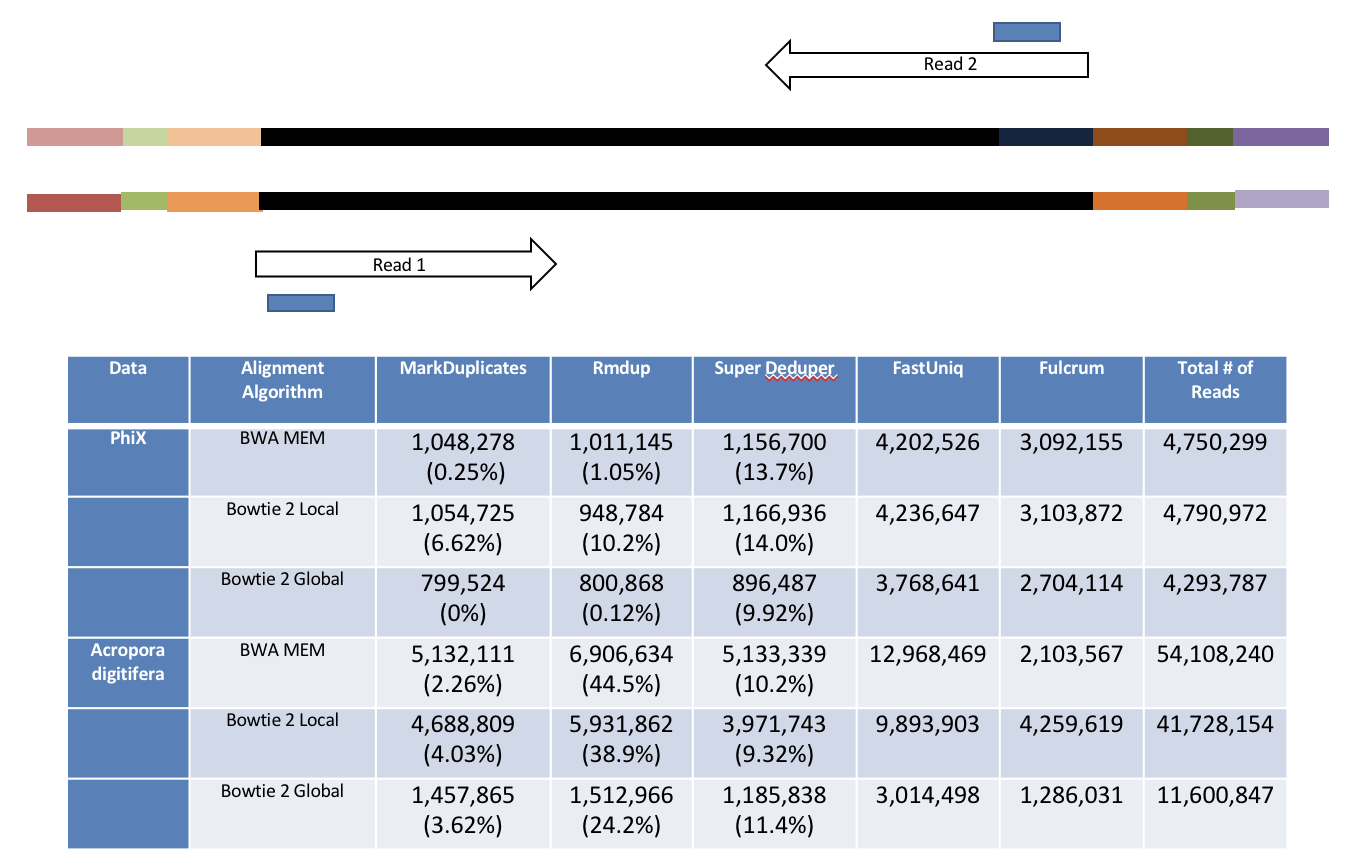
Super Deduper only uses a small portion of the reads to identify duplicates.
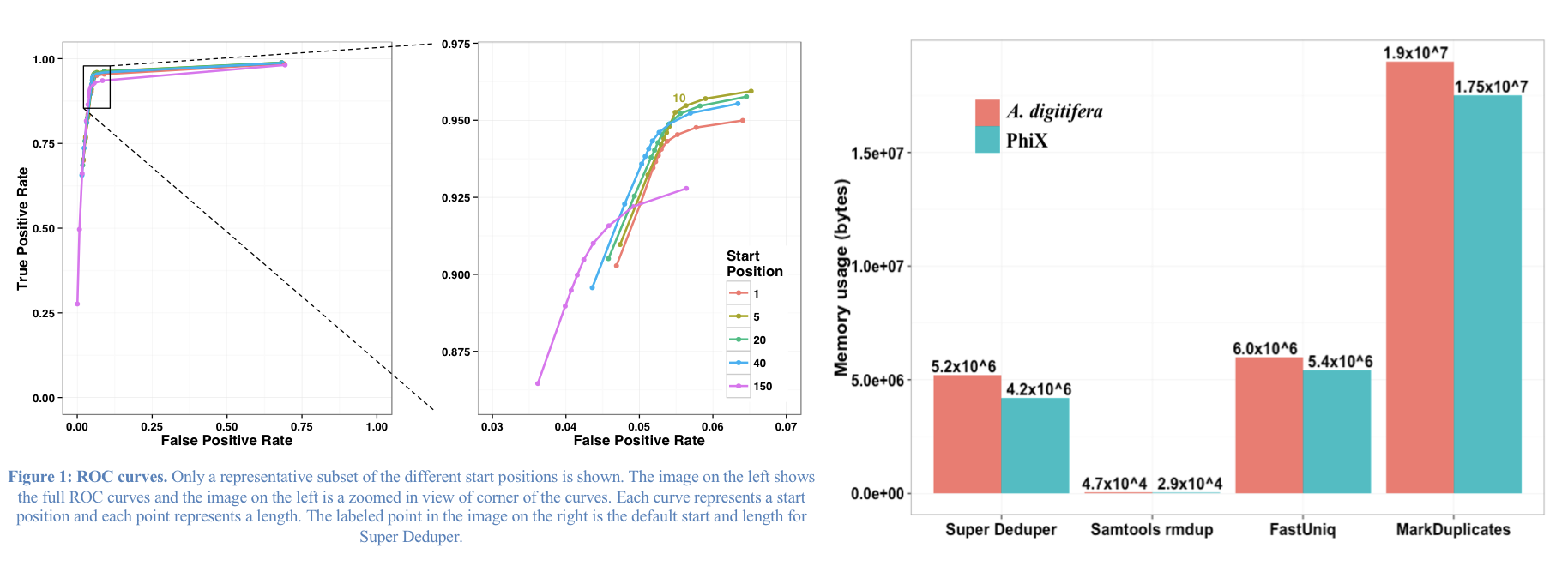
We calculated the Youden Index for every combination tested and the point that acquired the highest index value (as compared to Picard MarkDuplicates) occurred at a start position of 5bp and a length of 10bps (20bp total over both reads).
Adapter trimming by overlapping reads.
Consider the three scenarios below
Insert size > length of the number of cycles
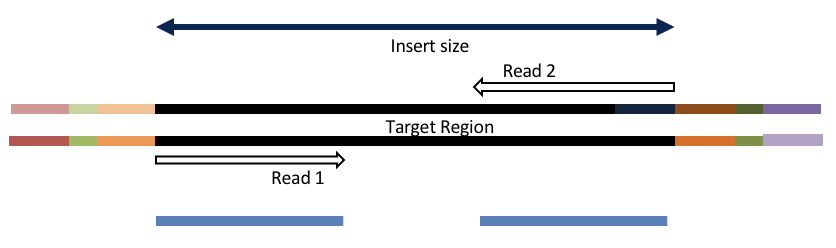
hts_AdapterTrimmer product: original pairs
hts_Overlapper product: original pairs
Insert size < length of the number of cycles (10bp min)
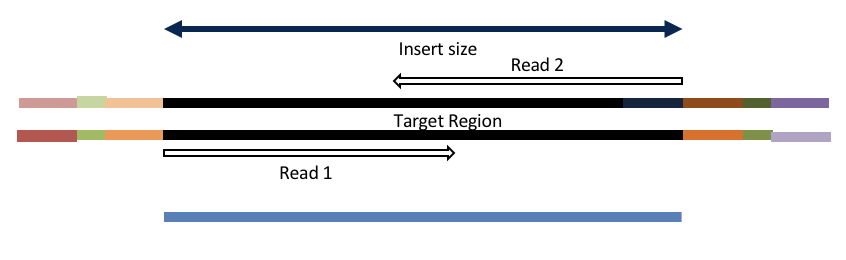
hts_AdapterTrimmer product: original pairs
hts_Overlapper product: extended, single
Insert size < length of the read length
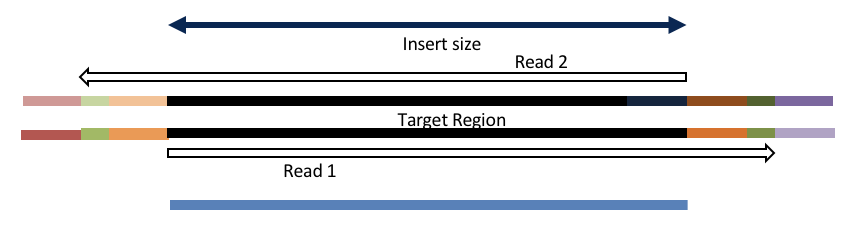
hts_AdapterTrimmer product: adapter trimmed, pairs
hts_Overlapper product: adapter trimmed, single
Both hts_AdapterTrimmer and hts_Overlapper employ this principle to identify and remove adapters for paired-end reads. For paired-end reads the difference between the two are the output, as overlapper produces single-end reads when the pairs overlap and adapter trimmer keeps the paired end format. For single-end reads, adapter trimmer identifies and removes adapters by looking for the adapter sequence, where overlapper just ignores single-end reads (nothing to overlap).
You can do a quick check for evidence of Illumina sequencing adapters using basic Linux commnads
Remember that Illumina reads must have P5 and P7 adapters and generally look like this (in R1 orientation):
P5---Index-Read1primer-------INSERT-------Read2primer--index--P7(rc)
|---R1 starts here-->
This sequence is P7(rc): ATCTCGTATGCCGTCTTCTGCTTG. It should present in any R1 that contains a full-length adapter sequence. It is easy to search for this sequence using zcat and grep:
cd /share/workshop/gwas_workshop/$USER/gwas_example/HTS_testing
zcat SL378587_subset_R1.fastq.gz | grep TCTCGTATGCCGTCTTCTGCTTG
Q-window trimming
As a sequencing run progresses the quality scores tend to get worse. Quality scores are essentially a guess about the accuracy of a base call, so it is common to trim of the worst quality bases.
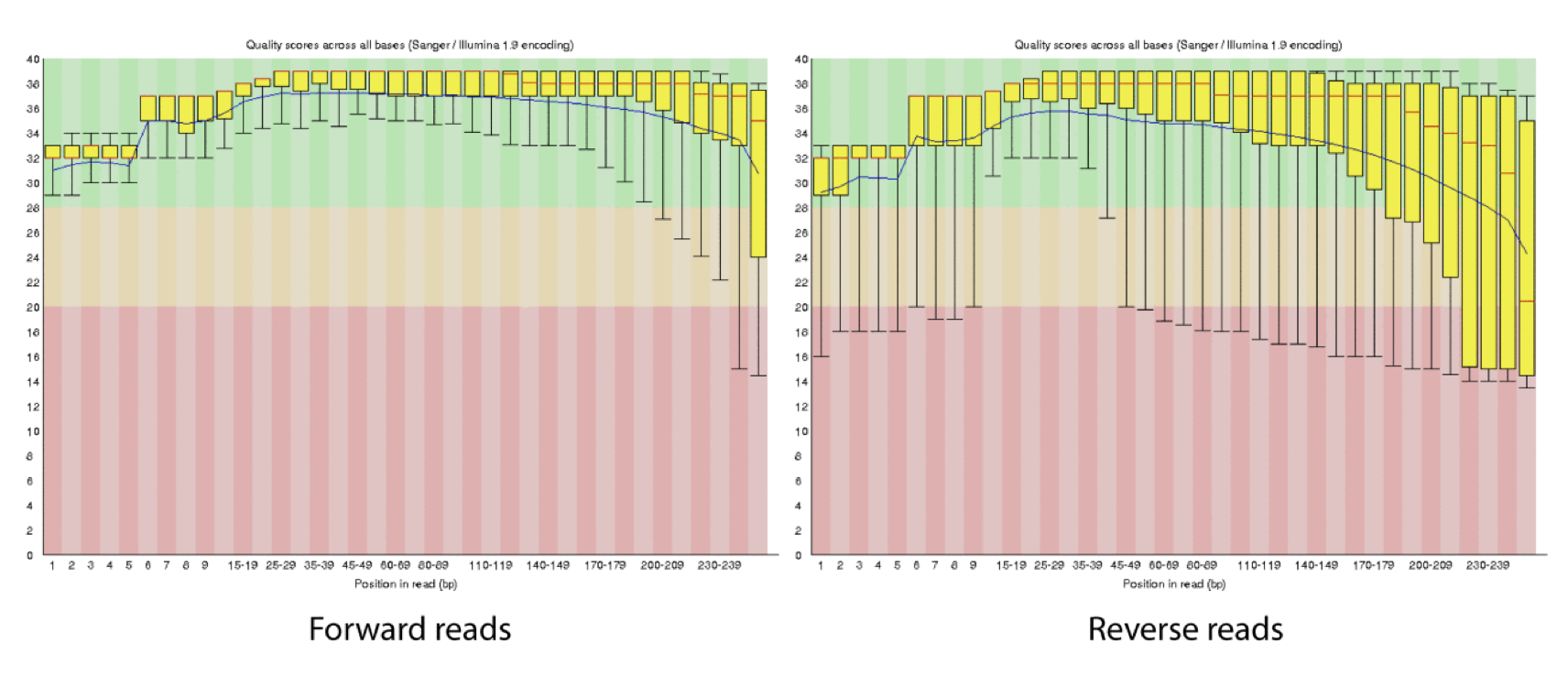
This is how reads commonly look, they start at “good” quality, increase to “excellent” and degrade to “poor”, with R2 always looking worse (except when they don’t) than R1 and get worse as the number of cycles increases.
hts_QWindowTrim trims 5’ and/or 3’ end of the sequence using a windowing (average quality in window) approach.
N Trimming
Bases that cannot be called are assigned an “N” by the Illumina base caller. These can be a problem for some applications, but most read mappers and quantification strategies should not be impacted unless N’s are frequent. By default, hts_NTrimmer will return the longest sequence that contains no Ns, but can also be configured to discard any reads containing Ns as well.
Lets put it all together
Start Group Exercise 2
cd /share/workshop/gwas_workshop/$USER/gwas_example/HTS_testing
hts_Stats -L SL378587_htsStats.json -N "initial stats" \
-1 SL378587_subset_R1.fastq.gz \
-2 SL378587_subset_R2.fastq.gz | \
hts_SeqScreener -A SL378587_htsStats.json -N "screen phix" | \
hts_SuperDeduper -A SL378587_htsStats.json -N "remove PCR duplicates" | \
hts_AdapterTrimmer -A SL378587_htsStats.json -N "trim adapters" | \
hts_QWindowTrim -A SL378587_htsStats.json -N "quality trim the ends of reads" | \
hts_NTrimmer -A SL378587_htsStats.json -N "remove any remaining 'N' characters" | \
hts_LengthFilter -A SL378587_htsStats.json -N "remove reads < 50bp" \
-n -m 50 | \
hts_Stats -A SL378587_htsStats.json -N "final stats" \
-f SL378587.htstream
Note the patterns:
- In the first routine we use -1 and -2 to specify the original reads.
- In the final routine -f fastq prefix to write out new preprocessed reads.
- For the log, we specify -L in the first app to write out to a new log, and then use -A for the second routine onward to append log output, generating a single log file at the end.
- All other parameters are algorithm specific, can review using –help
Questions
-
Review the final json output, how many reads do we have left?
-
Confirm that number by counting the number of reads in the final output files.
-
How many reads had adapters that were cut off?
-
How many PCR duplicates were there?
-
Anything else interesting?
Run HTStream on the Project.
We can now run the preprocessing routine across all samples on the real data using a SLURM script, hts_preproc.slurm, that we should take a look at now.
cd /share/workshop/gwas_workshop/$USER/gwas_example # We'll run this from the main directory
wget https://ucdavis-bioinformatics-training.github.io/2021-July-Genome-Wide-Association-Studies/software_scripts/scripts/hts_preproc.slurm
less hts_preproc.slurm
When you are done, type “q” to exit.
Double check to make sure that slurmout and 01-HTS_Preproc directories have been created for output, then after looking at the script, let’s run it.
cd /share/workshop/gwas_workshop/$USER/gwas_example
mkdir -p slurmout # -p tells mkdir not to complain if the directory already exists
mkdir -p 01-HTS_Preproc
sbatch -J hts.${USER} hts_preproc.slurm # moment of truth!
We can watch the progress of our task array using the ‘squeue’ command. Takes about 30 minutes to process each sample.
squeue -u $USER # use your username
End Group Exercise 2
Quality Assurance - Preprocessing statistics as QA/QC.
Beyond generating “better” data for downstream analysis, cleaning statistics also give you an idea as to the original quality and complexity of the sample, library generation, and sequencing quality.
This can help inform you of how you might change your procedures in the future, either sample preparation, or in library preparation.
I’ve found it best to perform QA/QC on both the run as a whole (poor samples can affect other samples) and on the samples themselves as they compare to other samples (BE CONSISTENT).
Reports such as Basespace for Illumina, are great ways to evaluate the run as a whole, the sequencing provider usually does this for you. PCA/MDS plots of the preprocessing summary are a great way to look for technical bias across your experiment. Poor quality samples often appear as outliers on the MDS plot and can ethically be removed due to identified technical issues.
Begin Group Exercise 3
-
Let’s make sure that all jobs completed successfully.
First check all the “htstream_*.out” and “htstream_*.err” files:
cd /share/workshop/gwas_workshop/$USER/gwas_example cat slurmout/htstream_*.outLook through the output and make sure you don’t see any errors. Now do the same for the err files:
cat slurmout/htstream_*.errAlso, check the output files. First check the number of forward and reverse output files (should be 22 each):
cd 01-HTS_Preproc ls */*R1* | wc -l ls */*R2* | wc -lDid you get the answer you expected, why or why not?
Check the sizes of the files as well. Make sure there are no zero or near-zero size files and also make sure that the size of the files are in the same ballpark as each other:
ls -lh * du -sh *IF for some reason HTStream didn’t finish, the files are corrupted or you missed the session, please let one of us know and we will help. You can also copy over the HTStream output.
cp -r /share/workshop/gwas_workshop/jli/gwas_example/01-HTS_Preproc /share/workshop/gwas_workshop/$USER/gwas_example/. -
Let’s take a look at the differences in adapter content between the input and output files. First look at the input file:
cd /share/workshop/gwas_workshop/$USER/gwas_example zless 00-RawData/SL378587_S1_L001_R1_001.fastq.gzLet’s search for the adapter sequence. Type ‘/’ (a forward slash), and then type AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC (the first part of the forward adapter). Press Enter. This will search for the sequence in the file and highlight each time it is found. You can now type “n” to cycle through the places where it is found. When you are done, type “q” to exit.
Now look at the output file:
zless 01-HTS_Preproc/SL378587/SL378587.htstream_R1.fastq.gzIf you scroll through the data (using the spacebar), you will see that some of the sequences have been trimmed. Now, try searching for AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC again. You shouldn’t find it (adapters were trimmed remember), but rarely is anything perfect. You may need to use Control-C to get out of the search and then “q” to exit the ‘less’ screen.
Lets grep for the sequence and get an idea of where it occurs in the raw sequences:
zcat 00-RawData/SL378587_S1_L001_R1_001.fastq.gz | grep --color=auto AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC- What do you observe? Are these sequences useful for analysis?
zcat 01-HTS_Preproc/SL378587/SL378587.htstream_R1.fastq.gz | grep --color=auto AGATCGGAAGAGCACACGTCTGAACTCCAGTCACLets grep for the sequence and count occurrences
zcat 00-RawData/SL378587_S1_L001_R1_001.fastq.gz | grep AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | wc -l zcat 01-HTS_Preproc/SL378587/SL378587.htstream_R1.fastq.gz | grep AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | wc -l-
What is the reduction in adapters found?
-
How could you modify the cleaning pipeline in order to remove the remaining sequences?
A MultiQC report for HTStream JSON files
Finally lets use MultiQC to generate a summary of our output. Currently MultiQC support for HTStream is in development by Bradley Jenner, and has not been included in the official MultiQC package. If you’d like to try it on your own data, you can find a copy here https://github.com/s4hts/MultiQC.
## Run multiqc to collect statistics and create a report:
cd /share/workshop/gwas_workshop/$USER/gwas_example
module load multiqc/htstream.dev0
mkdir -p 02-HTS_multiqc_report
multiqc -i HTSMultiQC-cleaning-report -o 02-HTS_multiqc_report ./01-HTS_Preproc
Transfer HTSMultiQC-cleaning-report_multiqc_report.html to your computer and open it in a web browser.
Or in case of emergency, download this copy: HTSMultiQC-cleaning-report_multiqc_report.html