
Project Setup
SNP arrays have been the most widely used genotyping technology in WGAS for decades, primarily due to their low costs. With the declining of sequencing cost, the switch to WGS is likely to be inevitable.
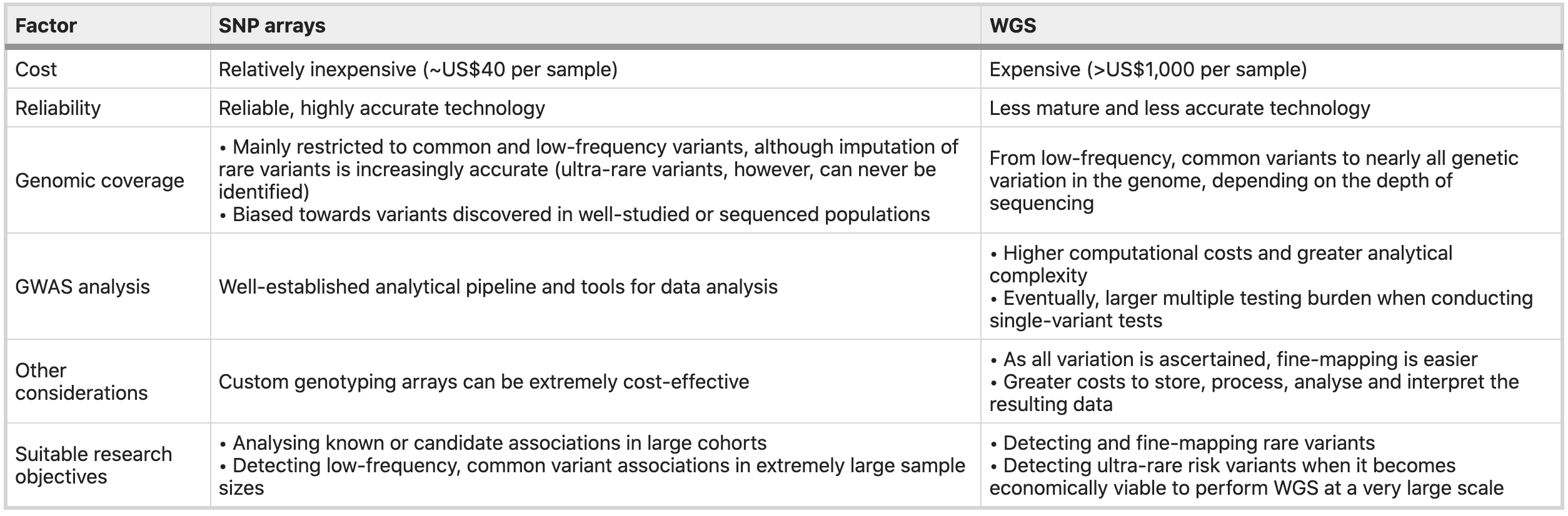
https://www.nature.com/articles/s41576-019-0127-1
For those who are interested in learning the process of using SNP array data for GWAS studies, please read this paper as a starting point: https://www.nature.com/articles/s41576-019-0127-1
In this workshop, we are going to focus on using WGS data.
Let’s set up a project directory for the week, and talk a bit about project philosophy..
Creating a Project Directory
First, create a directory for you and the example project in the workshop share directory:
cd
mkdir -p /share/workshop/gwas_workshop/$USER/gwas_example
Link raw fastq files
-
Next, go into that directory, create a raw data directory (we are going to call this 00-RawData) and cd into that directory. Lets then create symbolic links to the sample directories that contains the raw data.
cd /share/workshop/gwas_workshop/$USER/gwas_example mkdir 00-RawData cd 00-RawData/ cp /share/workshop/gwas_workshop/jli/gwas_example/00-RawData/* .This directory now contains the fastq files for all three samples that we are going to use.
-
Let’s create a sample sheet for the project and store sample names in a file called samples.txt
ls *_R1_* |awk 'BEGIN{FS="\\/"}{print $NF}' - |awk 'BEGIN{FS="_"}{print $1}' - > ../samples.txt cat ../samples.txt
Getting to know your data
-
Now, take a look at the raw data directory.
ls /share/workshop/gwas_workshop/$USER/gwas_example/00-RawDataLets get a better look at all the files in all of the directories.
ls -lah * -
View the contents of the files using the ‘less’ command, when gzipped used ‘zless’ (which is just the ‘less’ command for gzipped files):
zless SL378587_S1_L001_R1_001.fastq.gzMake sure you can identify which lines correspond to a read and which lines are the header, sequence, and quality values. Press ‘q’ to exit this screen.
-
Then, let’s figure out the number of reads in this file. A simple way to do that is to count the number of lines and divide by 4 (because the record of each read uses 4 lines). In order to do this use cat to output the uncompressed file and pipe that to “wc” to count the number of lines:
zcat SL378587_S1_L001_R1_001.fastq.gz | wc -lDivide this number by 4 and you have the number of reads in this file.
-
One more thing to try is to figure out the length of the reads without counting each nucleotide. First get the first 4 lines of the file (i.e. the first record):
zcat SL378587_S1_L001_R1_001.fastq.gz | head -2 | tail -1Note the header lines (1st and 3rd line) and sequence and quality lines (2nd and 4th) in each 4-line fastq block.
-
Then, copy and paste the DNA sequence line into the following command (replace [sequence] with the line):
echo -n [sequence] | wc -cThis will give you the length of the read. Also can do the bash one liner:
echo -n $(zcat SL378587_S1_L001_R1_001.fastq.gz | head -2 | tail -1) | wc -cSee if you can figure out how this command works.
This will give you the read count without doing any division. See if you can figure out how this command works:
zcat SL378587_S1_L001_R1_001.fastq.gz | grep -c "^@A00323:186"
Prepare our experiment folder for analysis
Now go back to your ‘gwas_example’ directory and create two directories called ‘slurmout’ and ‘01-HTS_Preproc’:
cd /share/workshop/gwas_workshop/$USER/gwas_example
mkdir References
mkdir slurmout
mkdir 01-HTS_Preproc
We’ll put reference sequence, genome, etc. in the References directory. The results of all our slurm script will output .out and .err files into the slurmout folder. The results of our preprocessing steps will be put into the 01-HTS_Preproc directory. The next step after that will go into a “02-…” directory, etc. You can collect scripts that perform each step, and notes and metadata relevant for each step, in the directory for that step. This way anyone looking to replicate your analysis has limited places to search for the commands you used. In addition, you may want to change the permissions on your original 00-RawData directory to “read only”, so that you can never accidentally corrupt (or delete) your raw data. We won’t worry about this here, because we’ve linked in sample folders.
Your directory should then look like the below:
$ ls
00-RawData 01-HTS_Preproc References samples.txt slurmout
Questions you should now be able to answer.
- How many reads are in the sample you checked?
- How many basepairs is R1, how many is R2?
- What is the name of the sequencer this dataset was run on?
- Which run number is this for that sequencer?
- What lane was this ran on?
- Randomly check the samples, were they all run on the same sequencer, run, and lane?