Gene ontology provides a controlled vocabulary for describing biological processes (BP ontology), molecular functions (MF ontology) and cellular components (CC ontology)
The GO ontologies themselves are organism-independent; terms are associated with genes for a specific organism through direct experimentation or through sequence homology with another organism and its GO annotation.
Terms are related to other terms through parent-child relationships in a directed acylic graph.
Enrichment analysis provides one way of drawing conclusions about a set of differential expression results.
1. topGO Example Using Kolmogorov-Smirnov Testing
Our first example uses Kolmogorov-Smirnov Testing for enrichment testing of our mouse DE results, with GO annotation obtained from the Bioconductor database org.Mm.eg.db.
The first step in each topGO analysis is to create a topGOdata object. This contains the genes, the score for each gene (here we use the p-value from the DE test), the GO terms associated with each gene, and the ontology to be used (here we use the biological process ontology)
infile<-"naive_v_memory.txt"tmp<-read.delim(infile)geneList<-tmp$P.Valuexx<-as.list(org.Mm.egENSEMBL2EG)names(geneList)<-xx[tmp$Gene.stable.ID]# Convert to entrezgene IDshead(geneList)
Annotated: number of genes (in our gene list) that are annotated with the term
Significant: n/a for this example, same as Annotated here
Expected: n/a for this example, same as Annotated here
raw.p.value: P-value from Kolomogorov-Smirnov test that DE p-values annotated with the term are smaller (i.e. more significant) than those not annotated with the term.
The Kolmogorov-Smirnov test directly compares two probability distributions based on their maximum distance.
To illustrate the KS test, we plot probability distributions of p-values that are and that are not annotated with the term GO:0048699 “generation of neurons” (1008 genes)m p-value 1.000. (This won’t exactly match what topGO does due to their elimination algorithm):
rna.pp.terms<-genesInTerm(GOdata)[["GO:0048699"]]# get genes associated with termp.values.in<-geneList[names(geneList)%in%rna.pp.terms]p.values.out<-geneList[!(names(geneList)%in%rna.pp.terms)]plot.ecdf(p.values.in,verticals=T,do.points=F,col="red",lwd=2,xlim=c(0,1),main="Empirical Distribution of DE P-Values by Annotation with 'generation of neurons'",cex.main=0.9,xlab="p",ylab="Probabilty(P-Value < p)")ecdf.out<-ecdf(p.values.out)xx<-unique(sort(c(seq(0,1,length=201),knots(ecdf.out))))lines(xx,ecdf.out(xx),col="black",lwd=2)legend("bottomright",legend=c("Genes Annotated with 'generation of neurons'","Other genes'"),lwd=2,col=2:1,cex=0.9)
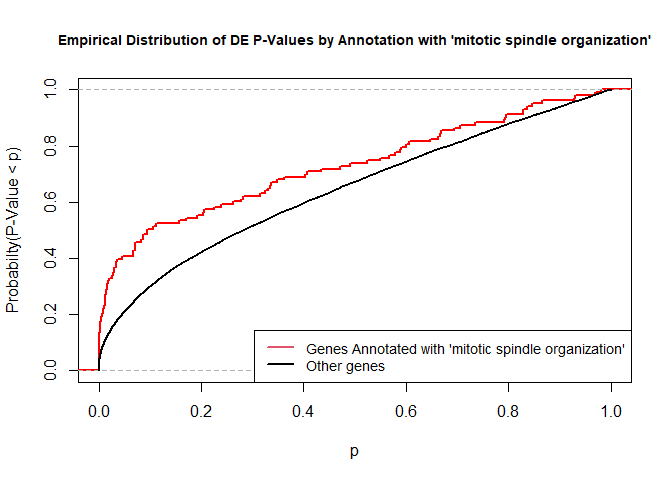
versus the probability distributions of p-values that are and that are not annotated with the term GO:0007052 “mitotic spindle organization” (103 genes) p-value 2.5x10-5.
rna.pp.terms<-genesInTerm(GOdata)[["GO:0007052"]]# get genes associated with termp.values.in<-geneList[names(geneList)%in%rna.pp.terms]p.values.out<-geneList[!(names(geneList)%in%rna.pp.terms)]plot.ecdf(p.values.in,verticals=T,do.points=F,col="red",lwd=2,xlim=c(0,1),main="Empirical Distribution of DE P-Values by Annotation with 'mitotic spindle organization'",cex.main=0.9,xlab="p",ylab="Probabilty(P-Value < p)")ecdf.out<-ecdf(p.values.out)xx<-unique(sort(c(seq(0,1,length=201),knots(ecdf.out))))lines(xx,ecdf.out(xx),col="black",lwd=2)legend("bottomright",legend=c("Genes Annotated with 'mitotic spindle organization'","Other genes"),lwd=2,col=2:1,cex=0.9)
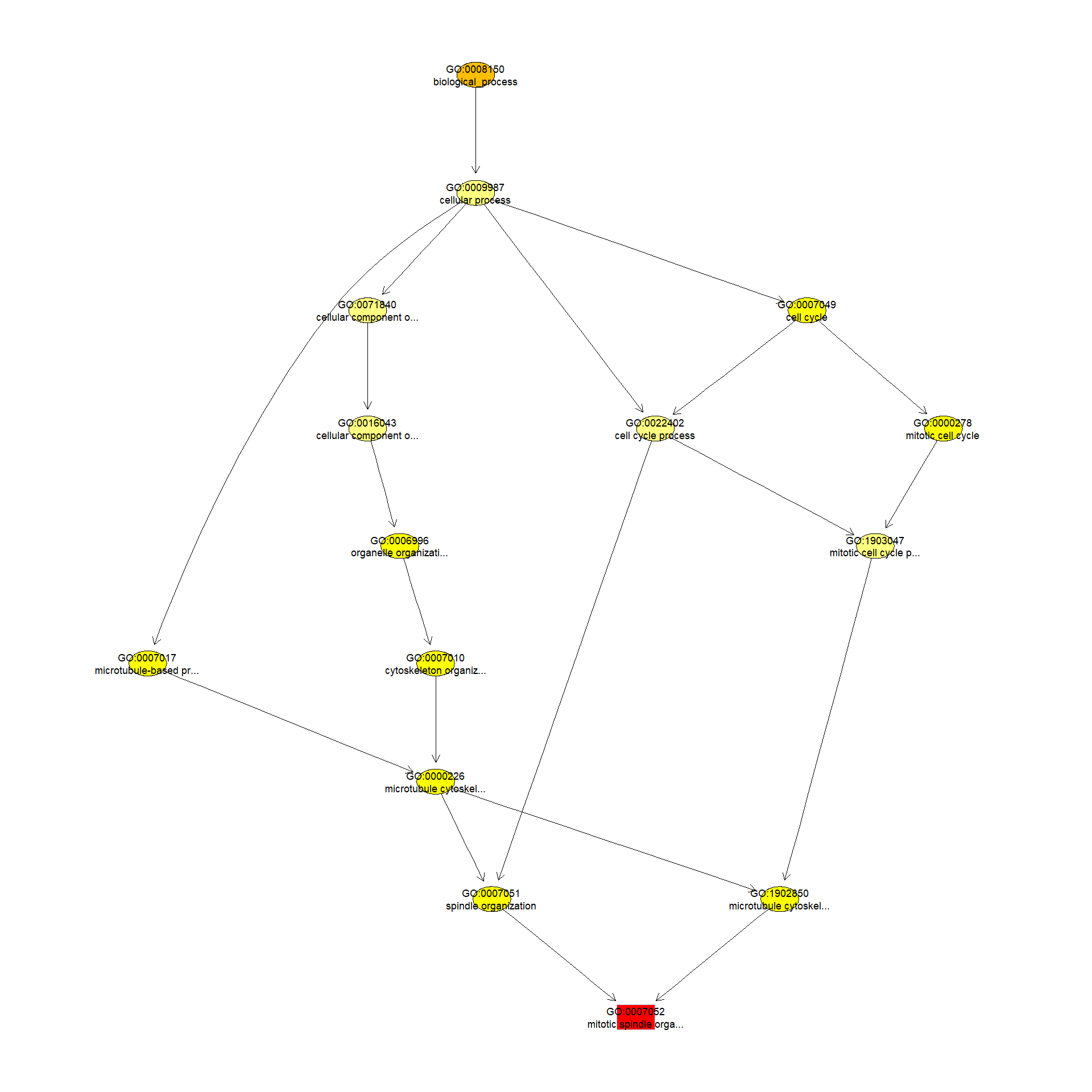
We can use the function showSigOfNodes to plot the GO graph for the most significant term and its parents, color coded by enrichment p-value (red is most significant):
Annotated: number of genes (in our gene list) that are annotated with the term
Significant: Number of significantly DE genes annotated with that term (i.e. genes where geneList = 1)
Expected: Under random chance, number of genes that would be expected to be significantly DE and annotated with that term
raw.p.value: P-value from Fisher’s Exact Test, testing for association between significance and pathway membership.
Fisher’s Exact Test is applied to the table:
Significance/Annotation
Annotated With GO Term
Not Annotated With GO Term
Significantly DE
n1
n3
Not Significantly DE
n2
n4
and compares the probability of the observed table, conditional on the row and column sums, to what would be expected under random chance.
Advantages over KS (or Wilcoxon) Tests:
Ease of interpretation
Easier directional testing
Disadvantages:
Relies on significant/non-significant dichotomy (an interesting gene could have an adjusted p-value of 0.051 and be counted as non-significant)
Less powerful
May be less useful if there are very few (or a large number of) significant genes
Quiz 1
##. KEGG Pathway Enrichment Testing With KEGGREST
KEGG, the Kyoto Encyclopedia of Genes and Genomes (https://www.genome.jp/kegg/), provides assignment of genes for many organisms into pathways.
We will access KEGG pathway assignments for mouse through the KEGGREST Bioconductor package, and then use some homebrew code for enrichment testing.
1. Get all mouse pathways and their genes:
# Pull all pathways for mmupathways.list<-keggList("pathway","mmu")head(pathways.list)
# Pull all genes for each pathwaypathway.codes<-sub("path:","",names(pathways.list))genes.by.pathway<-sapply(pathway.codes,function(pwid){pw<-keggGet(pwid)if(is.null(pw[[1]]$GENE))return(NA)pw2<-pw[[1]]$GENE[c(TRUE,FALSE)]pw2<-unlist(lapply(strsplit(pw2,split=";",fixed=T),function(x)x[1]))return(pw2)})head(genes.by.pathway)
2. Apply Wilcoxon rank-sum test to each pathway, testing if “in” p-values are smaller than “out” p-values:
# Wilcoxon test for each pathwaypVals.by.pathway<-t(sapply(names(genes.by.pathway),function(pathway){pathway.genes<-genes.by.pathway[[pathway]]list.genes.in.pathway<-intersect(names(geneList),pathway.genes)list.genes.not.in.pathway<-setdiff(names(geneList),list.genes.in.pathway)scores.in.pathway<-geneList[list.genes.in.pathway]scores.not.in.pathway<-geneList[list.genes.not.in.pathway]if(length(scores.in.pathway)>0){p.value<-wilcox.test(scores.in.pathway,scores.not.in.pathway,alternative="less")$p.value}else{p.value<-NA}return(c(p.value=p.value,Annotated=length(list.genes.in.pathway)))}))# Assemble output tableoutdat<-data.frame(pathway.code=rownames(pVals.by.pathway))outdat$pathway.name<-pathways.list[paste0("path:",outdat$pathway.code)]outdat$p.value<-pVals.by.pathway[,"p.value"]outdat$Annotated<-pVals.by.pathway[,"Annotated"]outdat<-outdat[order(outdat$p.value),]head(outdat,15)
p.value: P-value for Wilcoxon rank-sum testing, testing that p-values from DE analysis for genes in the pathway are smaller than those not in the pathway
Annotated: Number of genes in the pathway (regardless of DE p-value)
The Wilcoxon rank-sum test is the nonparametric analogue of the two-sample t-test. It compares the ranks of observations in two groups. It is more powerful than the Kolmogorov-Smirnov test for location tests.