
Last Updated: March 20 2022
Part 1: Loading data from CellRanger into R
Our first Markdown document concentrates on getting data into R and setting up our initial object.
Single Cell Analysis with Seurat and some custom code!
Seurat (now Version 4) is a popular R package that is designed for QC, analysis, and exploration of single cell data. Seurat aims to enable users to identify and interpret sources of heterogeneity from single cell transcriptomic measurements, and to integrate diverse types of single cell data. Further, the authors provide several tutorials, on their website.
The expression_data_cellranger.zip file contains the single cell matrix files and HDF5 files for four T cell samples from Mysore et al., 2021.
We start each markdown document with loading needed libraries for R:
# must have Seurat
library(Seurat)
library(kableExtra)
library(ggplot2)
Setup the experiment folder and data info
experiment_name = "Covid Example"
dataset_loc <- "./expression_data_cellranger"
ids <- c("conv_COVID", "conv_MMR", "conv_Tdap", "norm_COVID")
Read in the cellranger sample metrics csv files
Usually, there would be a single metrics summary file for each sample. In this case, we are working with a pool of samples that has been demultiplexed using antibody capture data prior to the workshop, and only have one metrics summary file generated for the entire pool.
The code in the box below reads in a single metrics summary file. To read in a summary file for each sample in your experiment, you should use the code in the next box instead.
experiment.metrics <- read.csv(file.path(dataset_loc, "metrics_summary.csv"))
sequencing.metrics <- data.frame(t(experiment.metrics[,c(1:19)]))
rownames(sequencing.metrics) <- gsub("\\.", " ", rownames(sequencing.metrics))
colnames(sequencing.metrics) <- "All samples"
sequencing.metrics %>%
kable(caption = 'Cell Ranger Results') %>%
pack_rows("Overview", 1, 3, label_row_css = "background-color: #666; color: #fff;") %>%
pack_rows("Sequencing Characteristics", 4, 9, label_row_css = "background-color: #666; color: #fff;") %>%
pack_rows("Mapping Characteristics", 10, 19, label_row_css = "background-color: #666; color: #fff;") %>%
kable_styling("striped")
| All samples | |
|---|---|
| Overview | |
| Estimated Number of Cells | 7,072 |
| Mean Reads per Cell | 89,005 |
| Median Genes per Cell | 1,880 |
| Sequencing Characteristics | |
| Number of Reads | 629,444,626 |
| Valid Barcodes | 94.4% |
| Sequencing Saturation | 84.0% |
| Q30 Bases in Barcode | 94.3% |
| Q30 Bases in RNA Read | 89.5% |
| Q30 Bases in UMI | 93.8% |
| Mapping Characteristics | |
| Reads Mapped to Genome | 90.0% |
| Reads Mapped Confidently to Genome | 85.4% |
| Reads Mapped Confidently to Intergenic Regions | 1.7% |
| Reads Mapped Confidently to Intronic Regions | 3.5% |
| Reads Mapped Confidently to Exonic Regions | 80.2% |
| Reads Mapped Confidently to Transcriptome | 76.5% |
| Reads Mapped Antisense to Gene | 1.7% |
| Fraction Reads in Cells | 75.5% |
| Total Genes Detected | 19,923 |
| Median UMI Counts per Cell | 5,671 |
d10x.metrics <- lapply(ids, function(i){
metrics <- read.csv(file.path(dataset_loc,paste0(i,"/outs"),"metrics_summary.csv"), colClasses = "character")
})
experiment.metrics <- do.call("rbind", d10x.metrics)
rownames(experiment.metrics) <- ids
sequencing.metrics <- data.frame(t(experiment.metrics[,c(1:19)]))
row.names(sequencing.metrics) <- gsub("\\."," ", rownames(sequencing.metrics))
sequencing.metrics %>%
kable(caption = 'Cell Ranger Results') %>%
pack_rows("Overview", 1, 3, label_row_css = "background-color: #666; color: #fff;") %>%
pack_rows("Sequencing Characteristics", 4, 9, label_row_css = "background-color: #666; color: #fff;") %>%
pack_rows("Mapping Characteristics", 10, 19, label_row_css = "background-color: #666; color: #fff;") %>%
kable_styling("striped")
Load the Cell Ranger Matrix Data and create the base Seurat object.
Cell Ranger provides a function cellranger aggr that will combine multiple samples into a single matrix file. However, when processing data in R this is unnecessary and we can quickly aggregate them in R.
Seurat provides a function Read10X and Read10X_h5 to read in 10X data folder. First we read in data from each individual sample folder.
Later, we initialize the Seurat object (CreateSeuratObject) with the raw (non-normalized data). Keep all cells with at least 200 detected genes. Also extracting sample names, calculating and adding in the metadata mitochondrial percentage of each cell. Adding in the metadata batchid and cell cycle. Finally, saving the raw Seurat object.
Load the Cell Ranger Matrix Data (hdf5 file) and create the base Seurat object.
d10x.data <- lapply(ids, function(i){
d10x <- Read10X_h5(file.path(dataset_loc, i, "/outs","raw_feature_bc_matrix.h5"))
colnames(d10x) <- paste(sapply(strsplit(colnames(d10x),split="-"),'[[',1L),i,sep="-")
d10x
})
names(d10x.data) <- ids
str(d10x.data)
If you don’t have the needed hdf5 libraries you can read in the matrix files like such
d10x.data <- sapply(ids, function(i){
d10x <- Read10X(file.path(dataset_loc, i, "/outs","raw_feature_bc_matrix"))
colnames(d10x) <- paste(sapply(strsplit(colnames(d10x), split="-"), '[[', 1L), i, sep="-")
d10x
})
names(d10x.data) <- ids
Lets recreate the barcode rank plot from the Cell Ranger web summary file.
In this case, we only have estimated cell counts for the pool as a whole. To color each barcode rank plot using the number of cells estimated by Cell Ranger, skip the code in the box below and use the next one instead.
plot_cellranger_cells <- function(ind){
xbreaks = c(1,1e1,1e2,1e3,1e4,1e5,1e6)
xlabels = c("1","10","100","1000","10k","100K","1M")
ybreaks = c(1,2,5,10,20,50,100,200,500,1000,2000,5000,10000,20000,50000,100000,200000,500000,1000000)
ylabels = c("1","2","5","10","2","5","100","2","5","1000","2","5","10k","2","5","100K","2","5","1M")
pl1 <- data.frame(index=seq.int(1,ncol(d10x.data[[ind]])),
nCount_RNA = sort(Matrix:::colSums(d10x.data[[ind]])+1,decreasing=T),
nFeature_RNA = sort(Matrix:::colSums(d10x.data[[ind]]>0)+1,decreasing=T)) %>%
ggplot() +
scale_color_manual(values=c("red2","blue4"), labels=c("Features", "UMI"), name=NULL) +
ggtitle(paste("CellRanger filltered cells:",ids[ind],sep=" ")) + xlab("Barcodes") + ylab("counts (UMI or Features") +
scale_x_continuous(trans = 'log2', breaks=xbreaks, labels = xlabels) +
scale_y_continuous(trans = 'log2', breaks=ybreaks, labels = ylabels) +
geom_line(aes(x=index, y=nCount_RNA, color = "UMI"), size=1.75) +
geom_line(aes(x=index, y=nFeature_RNA, color = "Features"), size=1.25)
return(pl1)
}
plot_cellranger_cells(1)
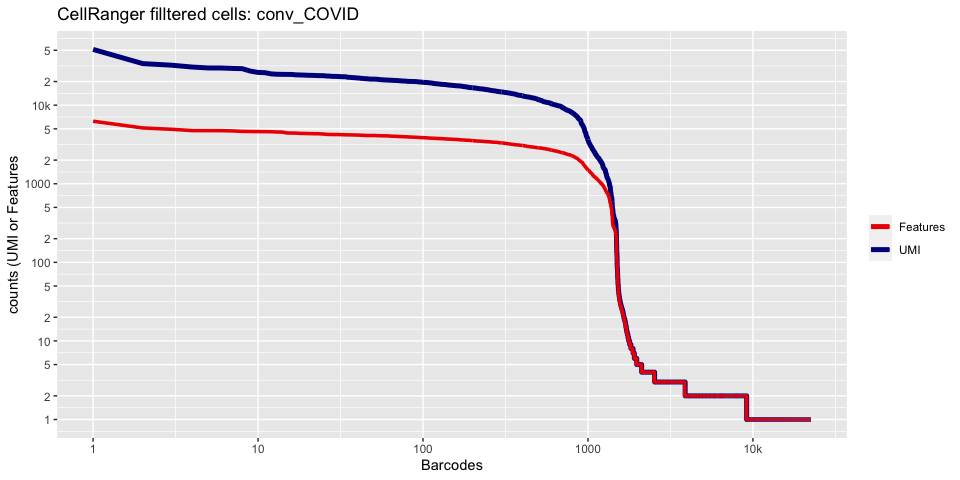
plot_cellranger_cells(2)
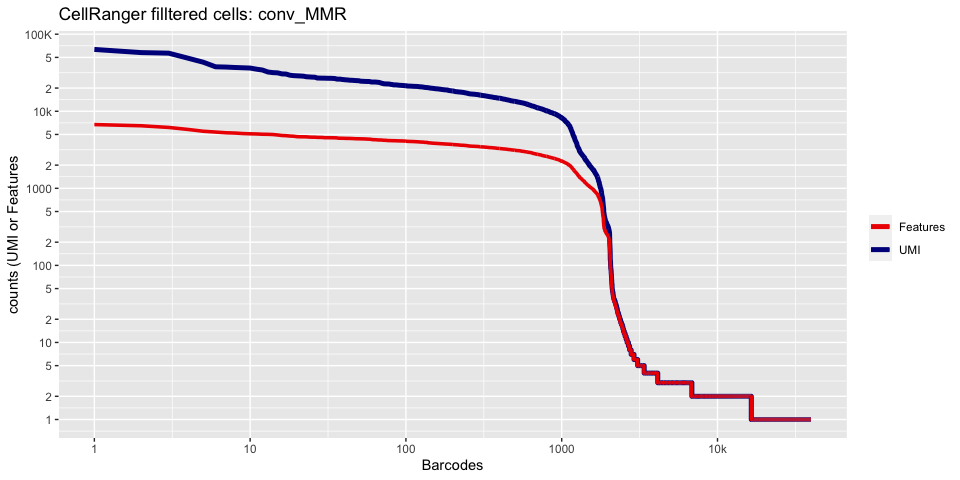
plot_cellranger_cells(3)
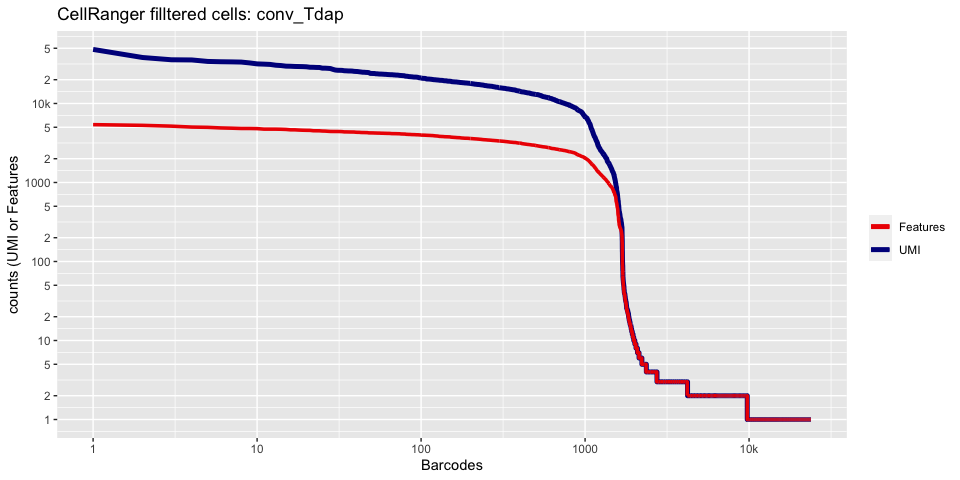
plot_cellranger_cells(4)
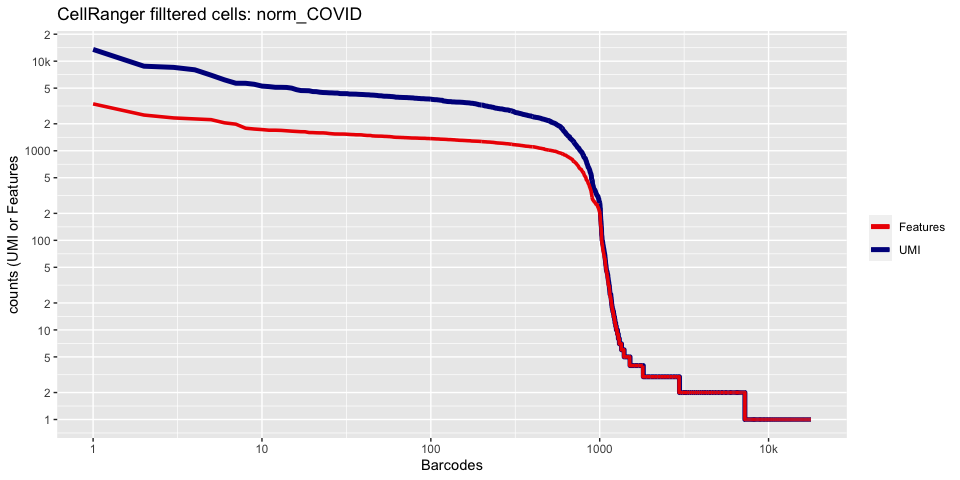
cr_filtered_cells <- as.numeric(gsub(",","",as.character(unlist(sequencing.metrics["Estimated Number of Cells",]))))
plot_cellranger_cells <- function(ind){
xbreaks = c(1,1e1,1e2,1e3,1e4,1e5,1e6)
xlabels = c("1","10","100","1000","10k","100K","1M")
ybreaks = c(1,2,5,10,20,50,100,200,500,1000,2000,5000,10000,20000,50000,100000,200000,500000,1000000)
ylabels = c("1","2","5","10","2","5","100","2","5","1000","2","5","10k","2","5","100K","2","5","1M")
pl1 <- data.frame(index=seq.int(1,ncol(d10x.data[[ind]])), nCount_RNA = sort(Matrix:::colSums(d10x.data[[ind]])+1,decreasing=T), nFeature_RNA = sort(Matrix:::colSums(d10x.data[[ind]]>0)+1,decreasing=T)) %>% ggplot() +
scale_color_manual(values=c("grey50","red2","blue4"), labels=c("UMI_Background", "Features", "UMI_Cells"), name=NULL) +
ggtitle(paste("CellRanger filltered cells:",ids[ind],sep=" ")) + xlab("Barcodes") + ylab("counts (UMI or Features") +
scale_x_continuous(trans = 'log2', breaks=xbreaks, labels = xlabels) +
scale_y_continuous(trans = 'log2', breaks=ybreaks, labels = ylabels) +
geom_line(aes(x=index, y=nCount_RNA, color=index<=cr_filtered_cells[ind] , group=1), size=1.75) +
geom_line(aes(x=index, y=nFeature_RNA, color="Features", group=1), size=1.25)
return(pl1)
}
plot_cellranger_cells(1)
plot_cellranger_cells(2)
plot_cellranger_cells(3)
plot_cellranger_cells(4)
Create the Seurat object
Filter criteria: remove genes that do not occur in a minimum of 0 cells and remove cells that don’t have a minimum of 200 features
experiment.data <- do.call("cbind", d10x.data)
experiment.aggregate <- CreateSeuratObject(
experiment.data,
project = experiment_name,
min.cells = 0,
min.features = 300,
names.field = 2,
names.delim = "\\-")
experiment.aggregate
str(experiment.aggregate)
The percentage of reads that map to the mitochondrial genome
- Low-quality / dying cells often exhibit extensive mitochondrial contamination.
- We calculate mitochondrial QC metrics with the PercentageFeatureSet function, which calculates the percentage of counts originating from a set of features.
- We use the set of all genes, in mouse these genes can be identified as those that begin with ‘mt’, in human data they begin with MT.
experiment.aggregate$percent.mito <- PercentageFeatureSet(experiment.aggregate, pattern = "^MT-")
summary(experiment.aggregate$percent.mito)
Lets spend a little time getting to know the Seurat object.
The Seurat object is the center of each single cell analysis. It stores all information associated with the dataset, including data, annotations, analyses, etc. The R function slotNames can be used to view the slot names within an object.
slotNames(experiment.aggregate)
head(experiment.aggregate[[]])
Question(s)
Finally, save the original object and view the object.
Original data set in Seurat class, with no filtering
save(experiment.aggregate,file="original_seurat_object.RData")
Get the next Rmd file
download.file("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2022-March-Single-Cell-RNA-Seq-Analysis/main/data_analysis/scRNA_Workshop-PART2.Rmd", "scRNA_Workshop-PART2.Rmd")
Session Information
sessionInfo()