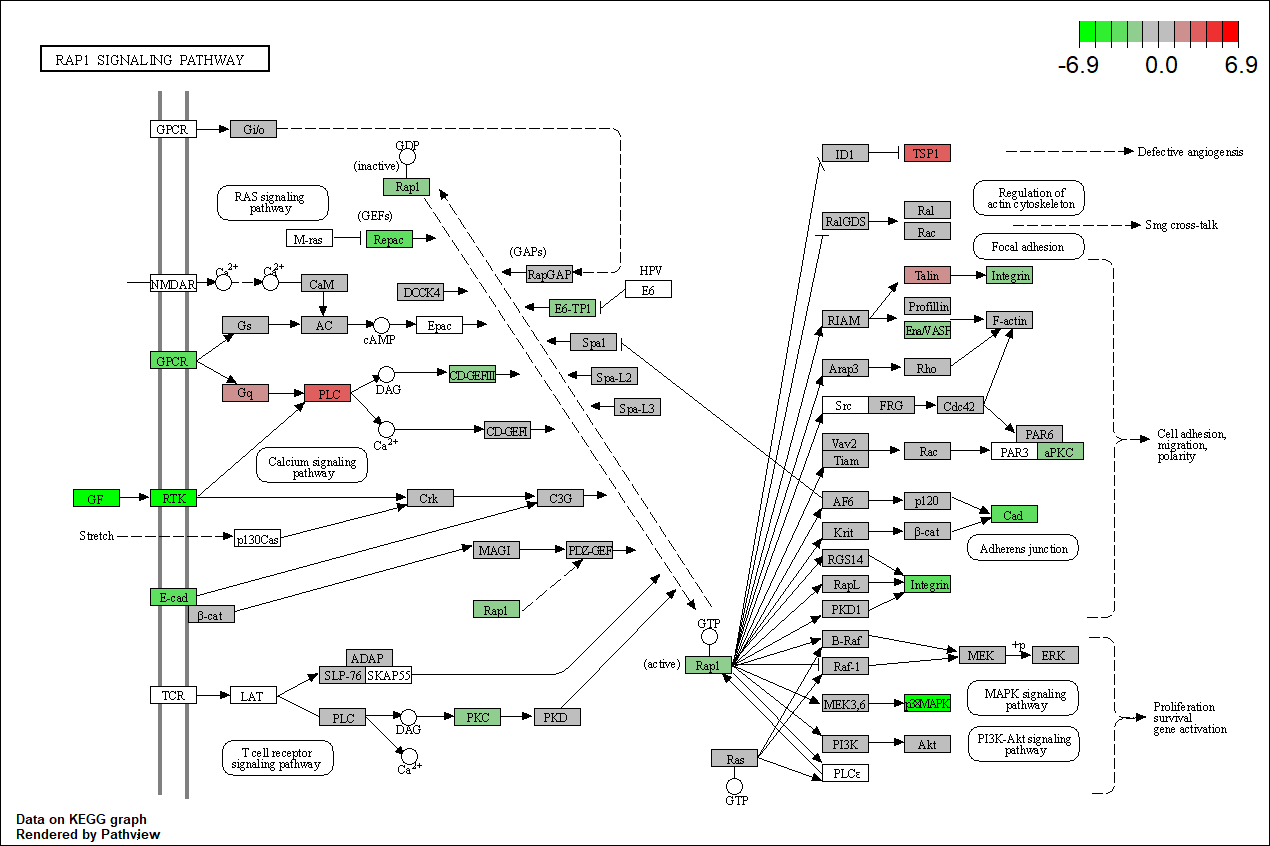
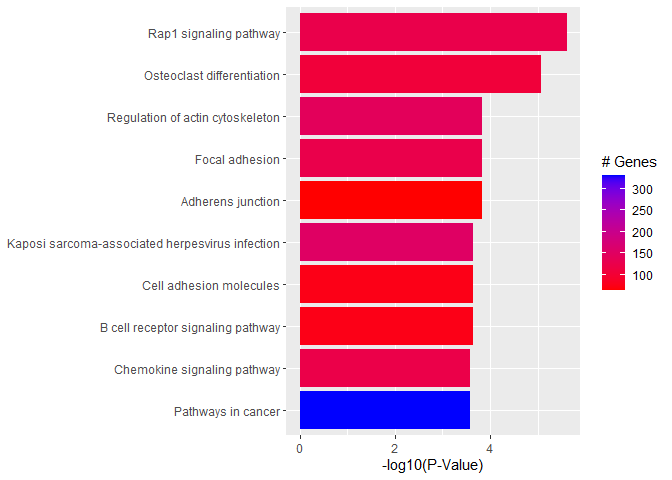
Files for examples were created in the DE analysis.
Gene Ontology (GO) Enrichment
Gene ontology provides a controlled vocabulary for describing biological processes (BP ontology), molecular functions (MF ontology) and cellular components (CC ontology)
The GO ontologies themselves are organism-independent; terms are associated with genes for a specific organism through direct experimentation or through sequence homology with another organism and its GO annotation.
Terms are related to other terms through parent-child relationships in a directed acylic graph.
Enrichment analysis provides one way of drawing conclusions about a set of differential expression results.
1. topGO Example Using Kolmogorov-Smirnov Testing
Our first example uses Kolmogorov-Smirnov Testing for enrichment testing of our mouse DE results, with GO annotation obtained from the Bioconductor database org.Mm.eg.db.
The first step in each topGO analysis is to create a topGOdata object. This contains the genes, the score for each gene (here we use the p-value from the DE test), the GO terms associated with each gene, and the ontology to be used (here we use the biological process ontology)
infile<-"WT.C_v_WT.NC.txt"DE<-read.delim(infile)## Add entrezgene IDs to top tabletmp<-bitr(DE$Gene.stable.ID,fromType="ENSEMBL",toType="ENTREZID",OrgDb=org.Mm.eg.db)
'select()' returned 1:many mapping between keys and columns
Warning in bitr(DE$Gene.stable.ID, fromType = "ENSEMBL", toType = "ENTREZID", :
5.55% of input gene IDs are fail to map...
id.conv<-subset(tmp,!duplicated(tmp$ENSEMBL))DE<-left_join(DE,id.conv,by=c("Gene.stable.ID"="ENSEMBL"))# Make gene listDE.nodupENTREZ<-subset(DE,!is.na(ENTREZID)&!duplicated(ENTREZID))geneList<-DE.nodupENTREZ$P.Valuenames(geneList)<-DE.nodupENTREZ$ENTREZIDhead(geneList)
topGO by default preferentially tests more specific terms, utilizing the topology of the GO graph. The algorithms used are described in detail here.
head(tab,15)
GO.ID Term
1 GO:0045071 negative regulation of viral genome replication
2 GO:0045944 positive regulation of transcription by RNA polymerase II
3 GO:0032731 positive regulation of interleukin-1 beta production
4 GO:0097202 activation of cysteine-type endopeptidase activity
5 GO:0045766 positive regulation of angiogenesis
6 GO:0051607 defense response to virus
7 GO:0032760 positive regulation of tumor necrosis factor production
8 GO:0045087 innate immune response
9 GO:0070374 positive regulation of ERK1 and ERK2 cascade
10 GO:0035458 cellular response to interferon-beta
11 GO:0001525 angiogenesis
12 GO:0051897 positive regulation of protein kinase B signaling
13 GO:2000117 negative regulation of cysteine-type endopeptidase activity
14 GO:0006002 fructose 6-phosphate metabolic process
15 GO:0031623 receptor internalization
Annotated Significant Expected raw.p.value
1 54 54 54 3.3e-09
2 784 784 784 3.8e-09
3 55 55 55 1.5e-08
4 25 25 25 8.4e-08
5 97 97 97 4.8e-07
6 242 242 242 6.1e-07
7 89 89 89 6.5e-07
8 591 591 591 7.9e-07
9 109 109 109 1.6e-06
10 44 44 44 2.0e-06
11 305 305 305 4.0e-06
12 61 61 61 5.0e-06
13 77 77 77 5.4e-06
14 10 10 10 5.4e-06
15 89 89 89 6.6e-06
Annotated: number of genes (in our gene list) that are annotated with the term
Significant: n/a for this example, same as Annotated here
Expected: n/a for this example, same as Annotated here
raw.p.value: P-value from Kolomogorov-Smirnov test that DE p-values annotated with the term are smaller (i.e. more significant) than those not annotated with the term.
The Kolmogorov-Smirnov test directly compares two probability distributions based on their maximum distance.
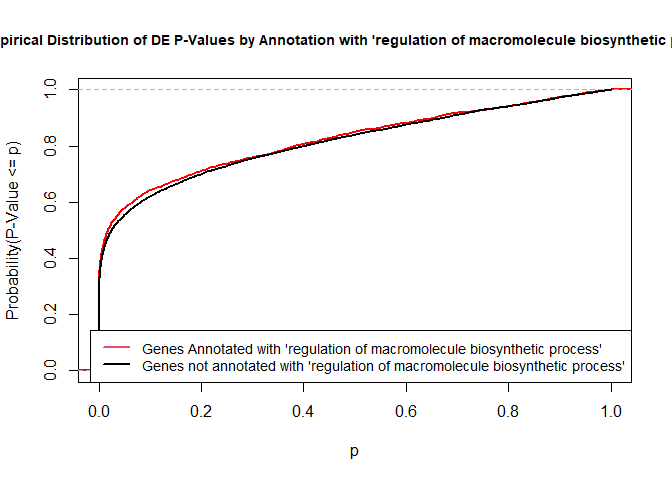
To illustrate the KS test, we plot probability distributions of p-values that are and that are not annotated with the term GO:0010556 “regulation of macromolecule biosynthetic process” (2344 genes) p-value 1.00. (This won’t exactly match what topGO does due to their elimination algorithm):
rna.pp.terms<-genesInTerm(GOdata)[["GO:0010556"]]# get genes associated with termp.values.in<-geneList[names(geneList)%in%rna.pp.terms]p.values.out<-geneList[!(names(geneList)%in%rna.pp.terms)]plot.ecdf(p.values.in,verticals=T,do.points=F,col="red",lwd=2,xlim=c(0,1),main="Empirical Distribution of DE P-Values by Annotation with 'regulation of macromolecule biosynthetic process'",cex.main=0.9,xlab="p",ylab="Probability(P-Value <= p)")ecdf.out<-ecdf(p.values.out)xx<-unique(sort(c(seq(0,1,length=201),knots(ecdf.out))))lines(xx,ecdf.out(xx),col="black",lwd=2)legend("bottomright",legend=c("Genes Annotated with 'regulation of macromolecule biosynthetic process'","Genes not annotated with 'regulation of macromolecule biosynthetic process'"),lwd=2,col=2:1,cex=0.9)
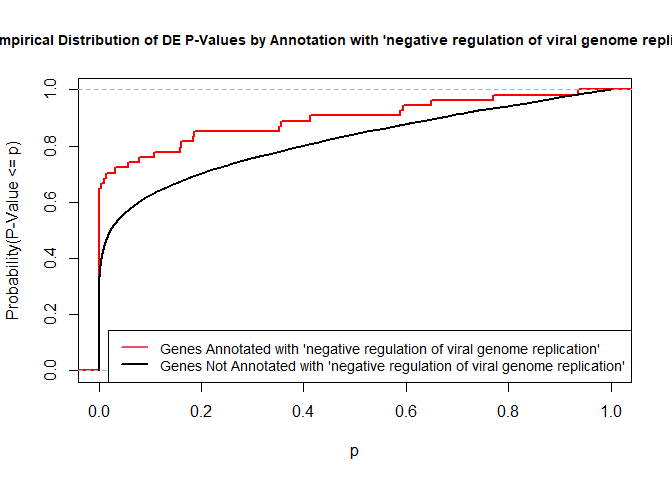
versus the probability distributions of p-values that are and that are not annotated with the term GO:0045071 “negative regulation of viral genome replication” (54 genes) p-value 3.3x10-9.
rna.pp.terms<-genesInTerm(GOdata)[["GO:0045071"]]# get genes associated with termp.values.in<-geneList[names(geneList)%in%rna.pp.terms]p.values.out<-geneList[!(names(geneList)%in%rna.pp.terms)]plot.ecdf(p.values.in,verticals=T,do.points=F,col="red",lwd=2,xlim=c(0,1),main="Empirical Distribution of DE P-Values by Annotation with 'negative regulation of viral genome replication'",cex.main=0.9,xlab="p",ylab="Probability(P-Value <= p)")ecdf.out<-ecdf(p.values.out)xx<-unique(sort(c(seq(0,1,length=201),knots(ecdf.out))))lines(xx,ecdf.out(xx),col="black",lwd=2)legend("bottomright",legend=c("Genes Annotated with 'negative regulation of viral genome replication'","Genes Not Annotated with 'negative regulation of viral genome replication'"),lwd=2,col=2:1,cex=0.9)
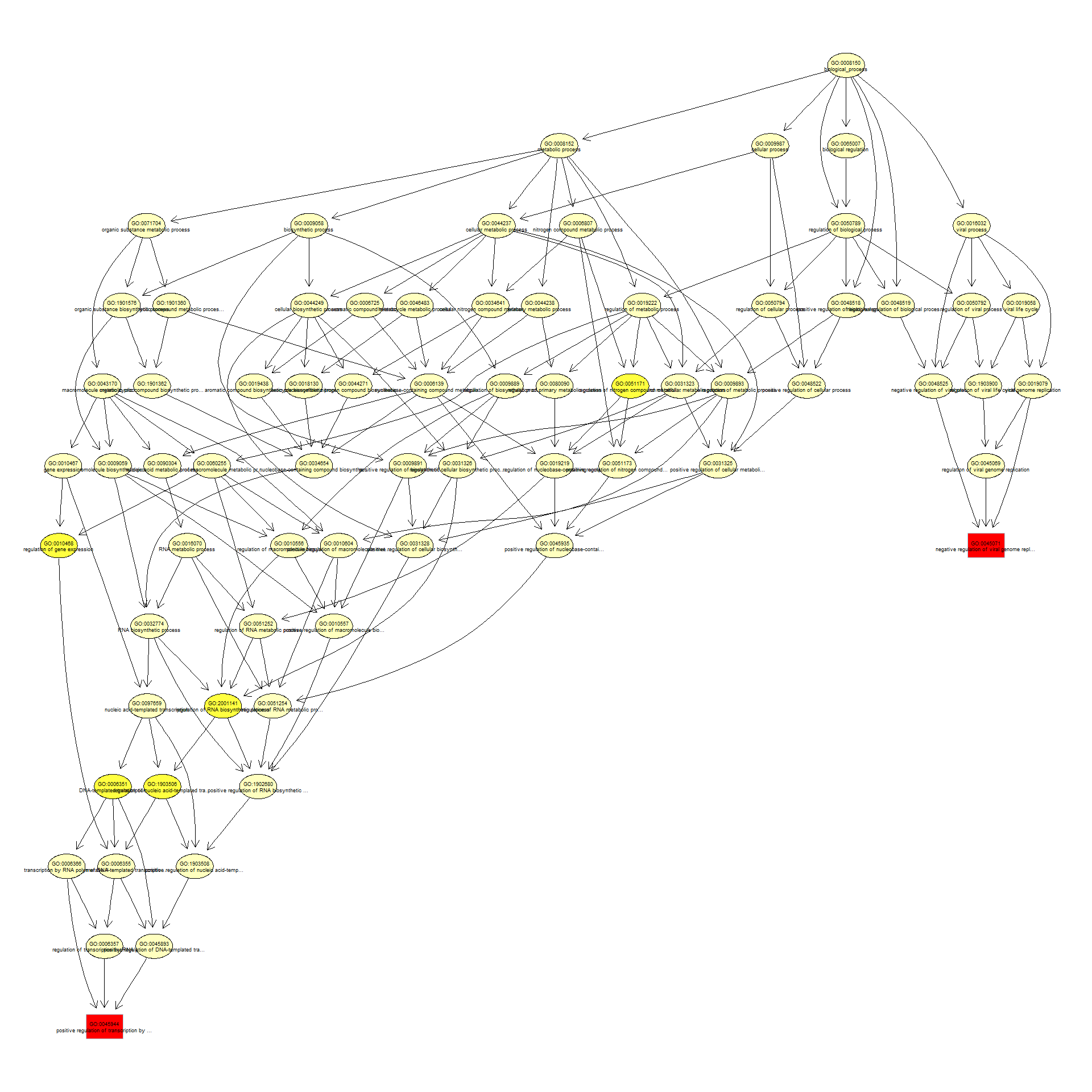
We can use the function showSigOfNodes to plot the GO graph for the 2 most significant terms and their parents, color coded by enrichment p-value (red is most significant):
Gene set enrichment analysis output includes the following columns:
setSize: Number of genes in pathway
enrichmentScore: GSEA enrichment score, a statistic reflecting the degree to which a pathway is overrepresented at the top or bottom of the gene list (the gene list here consists of the t-statistics from the DE test).