
Part 5: Enrichment, Model-Based DE, and Cell-Type Identification
Load libraries
library(Seurat)
library(ggplot2)
library(limma)
library(topGO)
Load the Seurat object
load("clusters_seurat_object.RData")
experiment.merged
## An object of class Seurat
## 21005 features across 10595 samples within 1 assay
## Active assay: RNA (21005 features, 5986 variable features)
## 3 dimensional reductions calculated: pca, tsne, umap
Idents(experiment.merged) <- "finalcluster"
1. Gene Ontology (GO) Enrichment of Genes Expressed in a Cluster
Gene Ontology provides a controlled vocabulary for describing gene products. Here we use enrichment analysis to identify GO terms that are overrepresented among the gene expressed in cells in a given cluster.
cluster12 <- subset(experiment.merged, idents = '12')
expr <- as.matrix(GetAssayData(cluster12))
# Filter out genes that are 0 for every cell in this cluster
bad <- which(rowSums(expr) == 0)
expr <- expr[-bad,]
# Select genes that are expressed > 0 in at least half of cells
n.gt.0 <- apply(expr, 1, function(x)length(which(x > 0)))
expressed.genes <- rownames(expr)[which(n.gt.0/ncol(expr) >= 0.5)]
all.genes <- rownames(expr)
# define geneList as 1 if gene is in expressed.genes, 0 otherwise
geneList <- ifelse(all.genes %in% expressed.genes, 1, 0)
names(geneList) <- all.genes
# Create topGOdata object
GOdata <- new("topGOdata",
ontology = "BP", # use biological process ontology
allGenes = geneList,
geneSelectionFun = function(x)(x == 1),
annot = annFUN.org, mapping = "org.Hs.eg.db", ID = "symbol")
# Test for enrichment using Fisher's Exact Test
resultFisher <- runTest(GOdata, algorithm = "elim", statistic = "fisher")
GenTable(GOdata, Fisher = resultFisher, topNodes = 20, numChar = 60)
## GO.ID Term Annotated Significant Expected Fisher
## 1 GO:0050861 positive regulation of B cell receptor signaling pathway 7 2 0.01 8.6e-05
## 2 GO:0072659 protein localization to plasma membrane 236 5 0.49 0.00010
## 3 GO:0050852 T cell receptor signaling pathway 125 4 0.26 0.00012
## 4 GO:0050727 regulation of inflammatory response 235 4 0.49 0.00127
## 5 GO:0021762 substantia nigra development 32 2 0.07 0.00196
## 6 GO:0000304 response to singlet oxygen 1 1 0.00 0.00207
## 7 GO:0042351 'de novo' GDP-L-fucose biosynthetic process 1 1 0.00 0.00207
## 8 GO:2000473 positive regulation of hematopoietic stem cell migration 1 1 0.00 0.00207
## 9 GO:0051623 positive regulation of norepinephrine uptake 1 1 0.00 0.00207
## 10 GO:0032745 positive regulation of interleukin-21 production 1 1 0.00 0.00207
## 11 GO:0072749 cellular response to cytochalasin B 1 1 0.00 0.00207
## 12 GO:1905475 regulation of protein localization to membrane 139 3 0.29 0.00284
## 13 GO:0034113 heterotypic cell-cell adhesion 45 2 0.09 0.00385
## 14 GO:1903615 positive regulation of protein tyrosine phosphatase activity 2 1 0.00 0.00414
## 15 GO:0031022 nuclear migration along microfilament 2 1 0.00 0.00414
## 16 GO:0021817 nucleokinesis involved in cell motility in cerebral cortex r... 2 1 0.00 0.00414
## 17 GO:0002728 negative regulation of natural killer cell cytokine producti... 2 1 0.00 0.00414
## 18 GO:0044855 plasma membrane raft distribution 2 1 0.00 0.00414
## 19 GO:1905430 cellular response to glycine 2 1 0.00 0.00414
## 20 GO:0001954 positive regulation of cell-matrix adhesion 48 2 0.10 0.00437
- Annotated: number of genes (out of all.genes) that are annotated with that GO term
- Significant: number of genes that are annotated with that GO term and meet our criteria for “expressed”
- Expected: Under random chance, number of genes that would be expected to be annotated with that GO term and meeting our criteria for “expressed”
- Fisher: (Raw) p-value from Fisher’s Exact Test
Quiz 1
Challenge Questions
If you have extra time:
- Rerun the enrichment analysis for the molecular function (MF) ontology.
- Think about how you write code to repeat the above enrichment analysis for every cluster (hint: ?base::sapply).
2. Model-based DE analysis in limma
limma is an R package for differential expression analysis of bulk RNASeq and microarray data. We apply it here to single cell data.
Limma can be used to fit any linear model to expression data and is useful for analyses that go beyond two-group comparisons. A detailed tutorial of model specification in limma is available here and in the limma User’s Guide.
# filter genes to those expressed in at least 10% of cells
keep <- rownames(expr)[which(n.gt.0/ncol(expr) >= 0.1)]
expr2 <- expr[keep,]
# Set up "design matrix" with statistical model
cluster12$proper.ident <- make.names(cluster12$orig.ident)
mm <- model.matrix(~0 + proper.ident + S.Score + G2M.Score + percent.mito + nFeature_RNA, data = cluster12[[]])
head(mm)
## proper.identA001.C.007 proper.identA001.C.104 proper.identB001.A.301 S.Score G2M.Score percent.mito nFeature_RNA
## AAACCCACAGAAGTTA_A001-C-007 1 0 0 0.016243559 -0.05567510 1.6666667 466
## AAACGCTAGGAGCAAA_A001-C-007 1 0 0 -0.059688163 0.01823853 2.0519836 626
## AAACGCTTCTCTGCTG_A001-C-007 1 0 0 0.244774911 0.74333121 1.1502030 1104
## AAAGAACCACGAAGAC_A001-C-007 1 0 0 -0.003307564 0.09956644 1.4218009 535
## AACAGGGGTCCCTGAG_A001-C-007 1 0 0 -0.035800756 0.01552573 0.7968127 744
## AAGGTAATCCTCAGAA_A001-C-007 1 0 0 -0.062652203 0.04392439 0.9419152 546
tail(mm)
## proper.identA001.C.007 proper.identA001.C.104 proper.identB001.A.301 S.Score G2M.Score percent.mito nFeature_RNA
## TTCCGTGTCCGCTGTT_B001-A-301 0 0 1 -0.08190609 -0.045416409 0.6387509 1064
## TTCTGTACATAGACTC_B001-A-301 0 0 1 0.02359308 -0.056658892 0.3048780 536
## TTCTTCCAGTCCCAAT_B001-A-301 0 0 1 0.02491281 -0.088061413 0.3700278 862
## TTGGATGCACGGTGCT_B001-A-301 0 0 1 -0.04342491 0.071349069 0.9578544 451
## TTTACGTGTGTCTTAG_B001-A-301 0 0 1 -0.09985271 -0.093126270 0.5780347 942
## TTTCGATAGACAACAT_B001-A-301 0 0 1 0.04867999 0.004383793 0.4915730 2371
# Fit model in limma
fit <- lmFit(expr2, mm)
head(coef(fit))
## proper.identA001.C.007 proper.identA001.C.104 proper.identB001.A.301 S.Score G2M.Score percent.mito nFeature_RNA
## CCNL2 0.2396261 -0.006494329 0.05299145 0.1177847 -0.3851735 0.099257545 0.0003433512
## CDK11A 0.6641791 0.089012473 0.09024772 -0.6450108 0.6608372 -0.013579988 0.0001919251
## GNB1 0.5498174 0.363893455 0.38644072 0.2396164 0.2658143 -0.001826895 0.0004433892
## SKI 0.2285806 0.398793577 -0.12774089 -0.1044831 -0.4867745 -0.059676145 0.0002570741
## KCNAB2 0.4760345 0.765881330 0.54504314 0.9751246 0.6724686 -0.103461311 -0.0001717929
## CAMTA1 -0.1310409 -0.095618782 0.07542118 -0.4560094 -0.4501812 -0.002074261 0.0004712436
# Test 'B001-A-301' - 'A001-C-007'
contr <- makeContrasts(proper.identB001.A.301 - proper.identA001.C.007, levels = colnames(coef(fit)))
contr
## Contrasts
## Levels proper.identB001.A.301 - proper.identA001.C.007
## proper.identA001.C.007 -1
## proper.identA001.C.104 0
## proper.identB001.A.301 1
## S.Score 0
## G2M.Score 0
## percent.mito 0
## nFeature_RNA 0
fit2 <- contrasts.fit(fit, contrasts = contr)
fit2 <- eBayes(fit2)
out <- topTable(fit2, n = Inf, sort.by = "P")
head(out, 30)
## logFC AveExpr t P.Value adj.P.Val B
## SLC26A2 3.0491797 1.0629482 17.062816 1.279092e-47 2.475042e-44 96.842841
## GUCA2A 1.6316995 0.4334461 11.246381 3.615387e-25 3.497887e-22 46.344851
## PHGR1 1.9817254 0.7501501 10.812613 1.263317e-23 8.148397e-21 42.872558
## XIST 1.3225033 0.3657630 10.380945 4.060678e-22 1.702613e-19 39.482858
## SLC26A3 1.8205559 0.6555777 10.370872 4.399517e-22 1.702613e-19 39.404586
## PDE3A 1.4273415 0.4332776 9.692689 8.833021e-20 2.848649e-17 34.228170
## CKB 2.0132899 1.4913849 8.459897 7.999548e-16 2.211304e-13 25.345494
## PIGR 1.9395745 1.3447380 8.150402 6.989238e-15 1.690522e-12 23.235771
## ATP1A1 1.4142129 0.6672922 7.771705 9.228991e-14 1.984233e-11 20.726518
## CLCA4 1.0457945 0.3366567 7.582066 3.258275e-13 6.304761e-11 19.501071
## MT-CO2 -1.5085157 2.7289681 -7.503861 5.447945e-13 9.583431e-11 19.001933
## MUC12 1.2542062 0.5705467 7.222223 3.364764e-12 5.425683e-10 17.235244
## TMSB4X 1.4108471 1.0055437 6.294871 9.469176e-10 1.409450e-07 11.778234
## FTH1 1.1423795 0.6892685 6.268453 1.102695e-09 1.519892e-07 11.631283
## CCND3 1.5327817 1.3641127 6.256938 1.178211e-09 1.519892e-07 11.567378
## S100A6 1.4073711 0.9677000 6.161607 2.031648e-09 2.457025e-07 11.041922
## CEACAM7 0.9032732 0.3321876 6.006647 4.859850e-09 5.531652e-07 10.201561
## NXPE1 1.0027720 0.4764160 5.894184 9.055717e-09 9.734896e-07 9.602475
## FKBP5 1.3994918 1.3421646 5.776481 1.720380e-08 1.752071e-06 8.985344
## FABP1 1.0951597 0.5854007 5.710409 2.455702e-08 2.375892e-06 8.643388
## HSP90AA1 -1.1122036 0.6331681 -5.655640 3.290375e-08 3.031846e-06 8.362388
## PIP4K2A 1.3017235 1.1825719 5.524949 6.555718e-08 5.766052e-06 7.700923
## PARP8 1.2093942 0.9387516 5.482825 8.164914e-08 6.869178e-06 7.490466
## SELENOP 0.8384054 0.3588361 5.438166 1.028959e-07 8.295982e-06 7.268813
## MUC13 0.9862040 0.5686166 5.423396 1.110395e-07 8.594456e-06 7.195839
## B2M 1.2463700 1.0201418 5.398320 1.263212e-07 9.211562e-06 7.072330
## NCL -0.8912419 0.4153055 -5.394937 1.285334e-07 9.211562e-06 7.055704
## RNF213 -1.3405337 1.4710884 -5.370101 1.459659e-07 1.008729e-05 6.933916
## FRYL 1.1859519 1.1869102 5.063632 6.749130e-07 4.503299e-05 5.470366
## TSPAN1 0.6987805 0.2775711 4.992235 9.543618e-07 6.155634e-05 5.139976
Output columns:
- logFC: log fold change (since we are working with Seurat’s natural log transformed data, will be natural log fold change)
- AveExpr: Average expression across all cells in expr2
- t: logFC divided by its standard error
- P.Value: Raw p-value (based on t) from test that logFC differs from 0
- adj.P.Val: Benjamini-Hochberg false discovery rate adjusted p-value
- B: log-odds that gene is DE
Quiz 2
BONUS: Cell type identification with scMRMA
scMRMA (single cell Multi-Resolution Marker-based Annotation Algorithm) classifies cells by iteratively clustering them then annotating based on a hierarchical external database.
The databases included with the current version are only for use with human and mouse, but a user-constructed hierarchichal database can be used.
The package can be installed from Github:
# Remove hashes to run
# install.packages("devtools")
# devtools::install_github("JiaLiVUMC/scMRMA")
suppressPackageStartupMessages(library(scMRMA))
result <- scMRMA(input = experiment.merged,
species = "Hs",
db = "panglaodb")
## Pre-defined cell type database panglaodb will be used.
## Multi Resolution Annotation Started.
## Level 1 annotation started.
## Level 2 annotation started.
## Level 3 annotation started.
## Level 4 annotation started.
## Uniform Resolution Annotation Started.
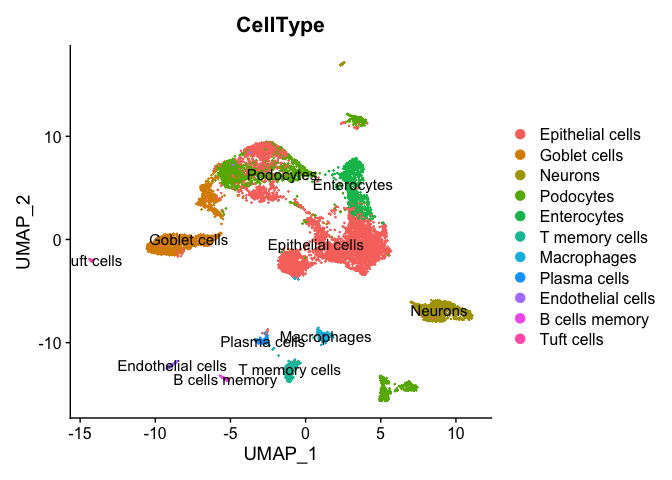
table(result$uniformR$annotationResult)
## UniformR
## Epithelial cells Goblet cells Neurons Podocytes Enterocytes T memory cells Macrophages Plasma cells Endothelial cells B cells memory Tuft cells
## 4556 1356 978 1992 819 329 195 142 99 80 49
## Add cell types to metadata
experiment.merged <- AddMetaData(experiment.merged, result$uniformR$annotationResult, col.name = "CellType")
table(experiment.merged$CellType, experiment.merged$orig.ident)
##
## A001-C-007 A001-C-104 B001-A-301
## Epithelial cells 202 1315 3039
## Goblet cells 56 361 939
## Neurons 936 10 32
## Podocytes 319 1292 381
## Enterocytes 0 20 799
## T memory cells 79 164 86
## Macrophages 94 64 37
## Plasma cells 75 44 23
## Endothelial cells 7 51 41
## B cells memory 4 49 27
## Tuft cells 2 46 1
table(experiment.merged$CellType, experiment.merged$finalcluster)
##
## 0 3 1 2 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 22 23 24 26
## Epithelial cells 1178 133 12 885 21 673 551 174 6 227 356 13 4 1 263 1 10 2 1 7 0 0 0 0 38
## Goblet cells 2 28 0 1 668 2 2 5 1 0 7 370 0 268 2 0 0 0 0 0 0 0 0 0 0
## Neurons 0 1 909 2 1 2 0 5 0 0 7 0 3 0 1 0 0 3 0 0 0 0 0 44 0
## Podocytes 2 757 1 1 0 1 117 452 1 1 25 5 0 0 5 269 199 0 156 0 0 0 0 0 0
## Enterocytes 5 0 0 2 0 1 0 4 626 177 0 0 1 3 0 0 0 0 0 0 0 0 0 0 0
## T memory cells 0 0 0 0 0 1 2 0 0 0 3 0 317 0 0 0 0 5 0 1 0 0 0 0 0
## Macrophages 0 0 0 4 0 2 0 0 0 0 0 0 3 0 0 0 0 186 0 0 0 0 0 0 0
## Plasma cells 0 0 0 0 0 0 1 0 0 0 1 1 1 0 0 0 0 0 0 138 0 0 0 0 0
## Endothelial cells 0 2 1 0 0 0 1 0 0 0 2 0 0 0 0 0 0 0 0 0 92 1 0 0 0
## B cells memory 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 79 0 0 0
## Tuft cells 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 49 0 0
DimPlot(experiment.merged, group.by = "CellType", label = TRUE)
Get the next Rmd file
download.file("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2022-December-Single-Cell-RNA-Seq-Analysis/main/data_analysis/scRNA_Workshop-PART6.Rmd", "scRNA_Workshop-PART6.Rmd")
Session Information
sessionInfo()
## R version 4.2.2 (2022-10-31)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Catalina 10.15.7
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] scMRMA_1.0 networkD3_0.4 data.tree_1.0.0 tidyr_1.2.1 RANN_2.6.1 plyr_1.8.8 irlba_2.3.5.1 Matrix_1.5-3 org.Hs.eg.db_3.16.0 topGO_2.50.0 SparseM_1.81 GO.db_3.16.0 AnnotationDbi_1.60.0 IRanges_2.32.0 S4Vectors_0.36.0 Biobase_2.58.0 graph_1.76.0 BiocGenerics_0.44.0 limma_3.54.0 ggplot2_3.4.0 SeuratObject_4.1.3
## [22] Seurat_4.3.0
##
## loaded via a namespace (and not attached):
## [1] igraph_1.3.5 lazyeval_0.2.2 sp_1.5-1 splines_4.2.2 listenv_0.8.0 scattermore_0.8 usethis_2.1.6 GenomeInfoDb_1.34.3 digest_0.6.30 htmltools_0.5.3 fansi_1.0.3 magrittr_2.0.3 memoise_2.0.1 tensor_1.5 cluster_2.1.4 ROCR_1.0-11 remotes_2.4.2 globals_0.16.2 Biostrings_2.66.0
## [20] matrixStats_0.63.0 spatstat.sparse_3.0-0 prettyunits_1.1.1 colorspace_2.0-3 blob_1.2.3 ggrepel_0.9.2 xfun_0.35 dplyr_1.0.10 callr_3.7.3 RCurl_1.98-1.9 crayon_1.5.2 jsonlite_1.8.4 progressr_0.11.0 spatstat.data_3.0-0 survival_3.4-0 zoo_1.8-11 glue_1.6.2 polyclip_1.10-4 gtable_0.3.1
## [39] zlibbioc_1.44.0 XVector_0.38.0 leiden_0.4.3 pkgbuild_1.4.0 future.apply_1.10.0 abind_1.4-5 scales_1.2.1 DBI_1.1.3 spatstat.random_3.0-1 miniUI_0.1.1.1 Rcpp_1.0.9 viridisLite_0.4.1 xtable_1.8-4 reticulate_1.26 bit_4.0.5 profvis_0.3.7 htmlwidgets_1.5.4 httr_1.4.4 RColorBrewer_1.1-3
## [58] ellipsis_0.3.2 ica_1.0-3 farver_2.1.1 urlchecker_1.0.1 pkgconfig_2.0.3 sass_0.4.4 uwot_0.1.14 deldir_1.0-6 utf8_1.2.2 labeling_0.4.2 tidyselect_1.2.0 rlang_1.0.6 reshape2_1.4.4 later_1.3.0 munsell_0.5.0 tools_4.2.2 cachem_1.0.6 cli_3.4.1 generics_0.1.3
## [77] RSQLite_2.2.19 devtools_2.4.5 ggridges_0.5.4 evaluate_0.18 stringr_1.4.1 fastmap_1.1.0 yaml_2.3.6 goftest_1.2-3 processx_3.8.0 fs_1.5.2 knitr_1.41 bit64_4.0.5 fitdistrplus_1.1-8 purrr_0.3.5 KEGGREST_1.38.0 pbapply_1.6-0 future_1.29.0 nlme_3.1-160 mime_0.12
## [96] compiler_4.2.2 rstudioapi_0.14 plotly_4.10.1 png_0.1-8 spatstat.utils_3.0-1 tibble_3.1.8 bslib_0.4.1 stringi_1.7.8 highr_0.9 ps_1.7.2 lattice_0.20-45 vctrs_0.5.1 pillar_1.8.1 lifecycle_1.0.3 spatstat.geom_3.0-3 lmtest_0.9-40 jquerylib_0.1.4 RcppAnnoy_0.0.20 bitops_1.0-7
## [115] data.table_1.14.6 cowplot_1.1.1 httpuv_1.6.6 patchwork_1.1.2 R6_2.5.1 promises_1.2.0.1 KernSmooth_2.23-20 gridExtra_2.3 parallelly_1.32.1 sessioninfo_1.2.2 codetools_0.2-18 pkgload_1.3.2 MASS_7.3-58.1 assertthat_0.2.1 withr_2.5.0 sctransform_0.3.5 GenomeInfoDbData_1.2.9 parallel_4.2.2 grid_4.2.2
## [134] rmarkdown_2.18 Rtsne_0.16 spatstat.explore_3.0-5 shiny_1.7.3