An object of class Seurat
11292 features across 6312 samples within 1 assay
Active assay: RNA (11292 features, 7012 variable features)
3 dimensional reductions calculated: pca, tsne, umap
Idents(experiment.merged)<-"finalcluster"
1. Gene Ontology (GO) Enrichment of Genes Expressed in a Cluster
Gene Ontology provides a controlled vocabulary for describing gene products. Here we use enrichment analysis to identify GO terms that are overrepresented among the gene expressed in cells in a given cluster.
cluster10<-subset(experiment.merged,idents='10')expr<-as.matrix(GetAssayData(cluster10))# Select genes that are expressed > 0 in at least half of cellsn.gt.0<-apply(expr,1,function(x)length(which(x>0)))expressed.genes<-rownames(expr)[which(n.gt.0/ncol(expr)>=0.5)]all.genes<-rownames(expr)# define geneList as 1 if gene is in expressed.genes, 0 otherwisegeneList<-ifelse(all.genes%in%expressed.genes,1,0)names(geneList)<-all.genes# Create topGOdata objectGOdata<-new("topGOdata",ontology="BP",# use biological process ontologyallGenes=geneList,geneSelectionFun=function(x)(x==1),annot=annFUN.org,mapping="org.Hs.eg.db",ID="symbol")# Test for enrichment using Fisher's Exact TestresultFisher<-runTest(GOdata,algorithm="elim",statistic="fisher")GenTable(GOdata,Fisher=resultFisher,topNodes=20,numChar=60)
GO.ID Term Annotated Significant Expected Fisher
1 GO:0030644 intracellular chloride ion homeostasis 8 4 0.32 0.00015
2 GO:0030050 vesicle transport along actin filament 15 5 0.59 0.00020
3 GO:0030036 actin cytoskeleton organization 462 40 18.19 0.00020
4 GO:0051660 establishment of centrosome localization 9 4 0.35 0.00025
5 GO:0006895 Golgi to endosome transport 17 5 0.67 0.00039
6 GO:0045053 protein retention in Golgi apparatus 5 3 0.20 0.00057
7 GO:0032534 regulation of microvillus assembly 5 3 0.20 0.00057
8 GO:0043268 positive regulation of potassium ion transport 19 5 0.75 0.00068
9 GO:0035176 social behavior 19 5 0.75 0.00068
10 GO:0009653 anatomical structure morphogenesis 1495 96 58.87 0.00093
11 GO:0030155 regulation of cell adhesion 473 33 18.62 0.00096
12 GO:2000650 negative regulation of sodium ion transmembrane transporter ... 6 3 0.24 0.00111
13 GO:0000381 regulation of alternative mRNA splicing, via spliceosome 42 7 1.65 0.00114
14 GO:0090630 activation of GTPase activity 82 10 3.23 0.00135
15 GO:1904908 negative regulation of maintenance of mitotic sister chromat... 2 2 0.08 0.00155
16 GO:0048669 collateral sprouting in absence of injury 2 2 0.08 0.00155
17 GO:0042060 wound healing 262 21 10.32 0.00155
18 GO:0007163 establishment or maintenance of cell polarity 160 15 6.30 0.00158
19 GO:0045944 positive regulation of transcription by RNA polymerase II 791 48 31.15 0.00158
20 GO:0040018 positive regulation of multicellular organism growth 14 4 0.55 0.00173
Annotated: number of genes (out of all.genes) that are annotated with that GO term
Significant: number of genes that are annotated with that GO term and meet our criteria for “expressed”
Expected: Under random chance, number of genes that would be expected to be annotated with that GO term and meeting our criteria for “expressed”
Fisher: (Raw) p-value from Fisher’s Exact Test
Quiz 1
2. Model-based DE analysis in limma
limma is an R package for differential expression analysis of bulk RNASeq and microarray data. We apply it here to single cell data.
Limma can be used to fit any linear model to expression data and is useful for analyses that go beyond two-group comparisons. A detailed tutorial of model specification in limma is available here and in the limma User’s Guide.
# filter genes to those expressed in at least 10% of cellskeep<-rownames(expr)[which(n.gt.0/ncol(expr)>=0.1)]expr2<-expr[keep,]# Set up "design matrix" with statistical modelcluster10$proper.ident<-make.names(cluster10$orig.ident)mm<-model.matrix(~0+proper.ident+S.Score+G2M.Score+percent_MT+nFeature_RNA,data=cluster10[[]])head(mm)
# Test 'B001-A-301' - 'A001-C-007'contr<-makeContrasts(proper.identB001.A.301-proper.identA001.C.007,levels=colnames(coef(fit)))fit2<-contrasts.fit(fit,contrasts=contr)fit2<-eBayes(fit2)out<-topTable(fit2,n=Inf,sort.by="P")head(out,30)
# load gene set preparation functionsource("https://raw.githubusercontent.com/IanevskiAleksandr/sc-type/master/R/gene_sets_prepare.R")# load cell type annotation functionsource("https://raw.githubusercontent.com/IanevskiAleksandr/sc-type/master/R/sctype_score_.R")
Read in marker database:
# DB filedb_="https://raw.githubusercontent.com/IanevskiAleksandr/sc-type/master/ScTypeDB_full.xlsx";tissue="Intestine"# e.g. Immune system,Pancreas,Liver,Eye,Kidney,Brain,Lung,Adrenal,Heart,Intestine,Muscle,Placenta,Spleen,Stomach,Thymus # prepare gene setsgs_list=gene_sets_prepare(db_,tissue)
Let’s take a look at the structure of the marker database:
# get cell-type by cell matrixscale.data<-GetAssayData(experiment.merged,"scale")es.max=sctype_score(scRNAseqData=scale.data,scaled=TRUE,gs=gs_list$gs_positive,gs2=gs_list$gs_negative)# cell type scores for first celles.max[,1]
The ScType developer suggests that assignments with a score less than (number of cells in cluster)/4 are low confidence and should be set to unknown:
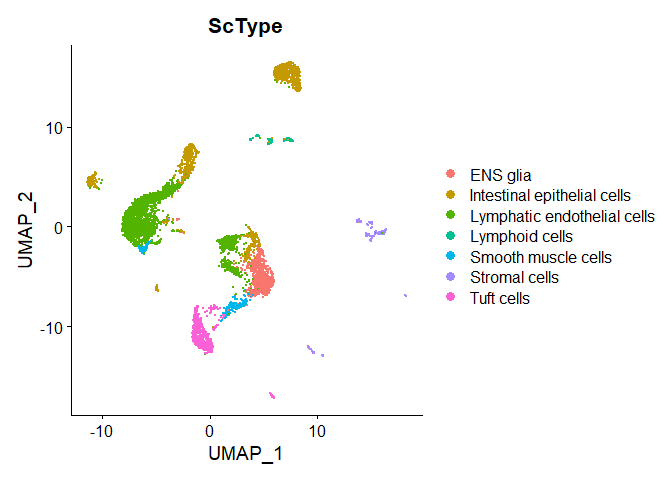
tmp<-data.frame(cell=colnames(experiment.merged),finalcluster=experiment.merged$finalcluster)tmp<-left_join(tmp,cL_results.top1,by="finalcluster")# set assignments with scores less than ncells/4 to unknowntmp$ScType.filtered<-ifelse(tmp$scores<tmp$ncells/4,"Unknown",tmp$ScType)experiment.merged$ScType.filtered<-tmp$ScType.filteredDimPlot(experiment.merged,group.by="ScType.filtered")