
BASE R GRAPHICS
Download and open the Rmd file for this section:
download.file("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2025-August-Intermediate-Visualization-for-Bioinformatics/refs/heads/master/R/baseRgraphics.Rmd", "baseRgrapics.Rmd")
First let’s download some data to work with and take a look at it:
counts = read.delim("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2025-August-Intermediate-Visualization-for-Bioinformatics/refs/heads/master/R/raw_counts.txt", header=T)
head(counts)
## C61 C62 C63 C64 C91 C92 C93 C94 I561 I562 I563 I564 I591 I592
## AT1G01010 322 346 256 396 372 506 361 342 638 488 440 479 770 430
## AT1G01020 149 87 162 144 189 169 147 108 163 141 119 147 182 156
## AT1G01030 15 32 35 22 24 33 21 35 18 8 54 35 23 8
## AT1G01040 687 469 568 651 885 978 794 862 799 769 725 715 811 567
## AT1G01046 1 1 5 4 5 3 0 2 4 3 1 0 2 8
## AT1G01050 1447 1032 1083 1204 1413 1484 1138 938 1247 1516 984 1044 1374 1355
## I593 I594 I861 I862 I863 I864 I891 I892 I893 I894
## AT1G01010 656 467 143 453 429 206 567 458 520 474
## AT1G01020 153 177 43 144 114 50 161 195 157 144
## AT1G01030 16 24 42 17 22 39 26 28 39 30
## AT1G01040 831 694 345 575 605 404 735 651 725 591
## AT1G01046 8 1 0 4 0 3 5 7 0 5
## AT1G01050 1437 1577 412 1338 1051 621 1434 1552 1248 1186
Let’s start with a simple plot with axis labels:
plot(counts[,"C61"], counts[,"C62"], xlab="C61", ylab="C62")
Let’s add a x=y line:
plot(counts[,"C61"], counts[,"C62"], xlab="C61", ylab="C62")
abline(0,1)
Change the plotting character:
plot(counts[,"C61"], counts[,"C62"], xlab="C61", ylab="C62", pch=23, col="red")
abline(0,1)
Create your own axes:
plot(counts[,"C61"], counts[,"C62"], xlab="C61", ylab="C62", pch=23, axes=F)
axis(1, at=seq(0,80000,by=10000))
axis(2, at=seq(0,100000,by=10000))
Add color to various things and change axis line widths:
plot(counts[,"C61"], counts[,"C62"], xlab="C61", ylab="C62", pch=23, axes=F, col="red")
axis(1, at=seq(0,80000,by=10000), col="blue", col.ticks="orange", col.axis="green", lwd=5)
axis(2, at=seq(0,100000,by=10000), col="blue", col.ticks="orange", col.axis="green", lwd=5)
Specify the ticks in a different way:
# xaxp=c(start,end,number of intervals)
plot(counts[,"C61"], counts[,"C62"], xlab="C61", ylab="C62",
xaxp=c(0,80000,4),
yaxp=c(0,100000,4))
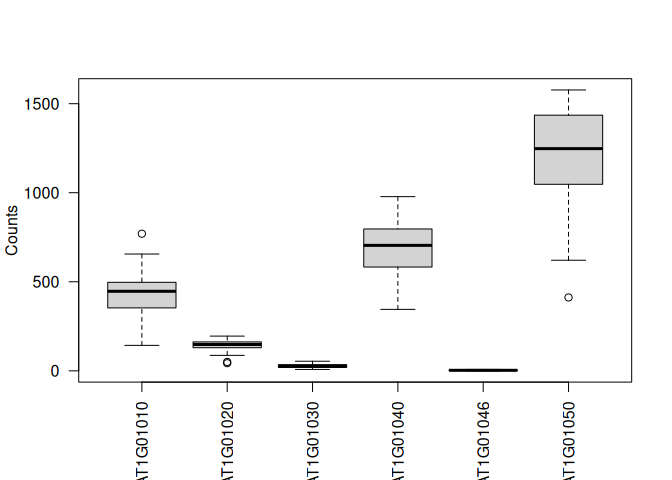
Now, a simple boxplot:
boxplot(counts)
Let’s create a smaller dataset to work with, add some factor data, and then use the melt function to change the shape of our data:
library(reshape2)
tc = as.data.frame(t(counts))
regions = c(rep("region1",6), rep("region2",6), rep("region3",6), rep("region4",6))
tc2 = tc[,1:6]
tc2$regions = regions
tcm = melt(tc2)
## Using regions as id variables
head(tcm)
## regions variable value
## 1 region1 AT1G01010 322
## 2 region1 AT1G01010 346
## 3 region1 AT1G01010 256
## 4 region1 AT1G01010 396
## 5 region1 AT1G01010 372
## 6 region1 AT1G01010 506
Now create a boxplot with colors and with the data separated by region:
cols <- c("red", "blue", "orange", "green")
boxplot(value ~ regions, data=tcm, col = cols, xlab="GeneIDs", ylab="Counts")
We can also add the points to the boxplot:
cols <- c("red", "blue", "orange", "green")
boxplot(value ~ regions, data=tcm, col = cols, xlab="GeneIDs", ylab="Counts")
stripchart(value ~ regions, data=tcm,
vertical=TRUE,
method="jitter",
pch=19,
add=TRUE)
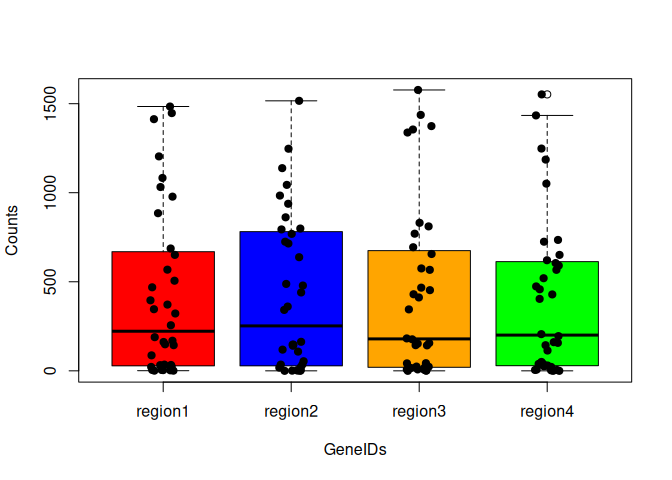
Something a little more complicated. Boxplots of regions vs. values, but separated by variable (GeneIDs):
boxplot(value ~ regions + variable, data=tcm, col = cols, xaxt="n", yaxt="n", xlab="GeneIDs", ylab="Counts")
# use the par function to get the dimensions of the plot
xlen = par("usr")[2]
axis(side=1, labels=colnames(tc2)[1:6], at=seq(2,xlen-2,length.out=6), cex.axis=0.7)
axis(side=2, las=2)
# add a legend
legend("topleft", fill=cols, legend=unique(regions))
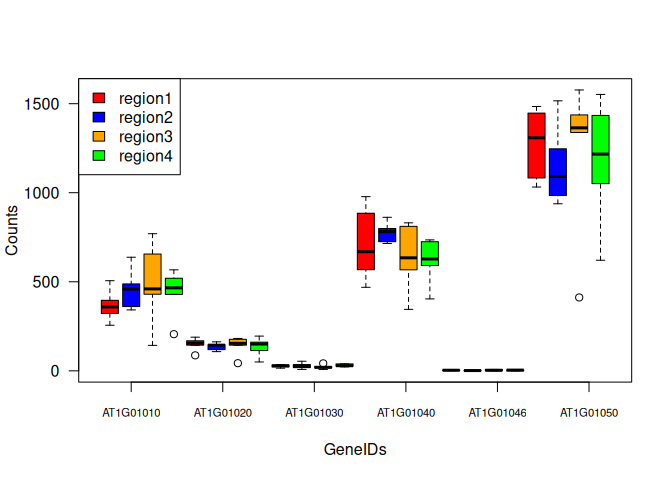
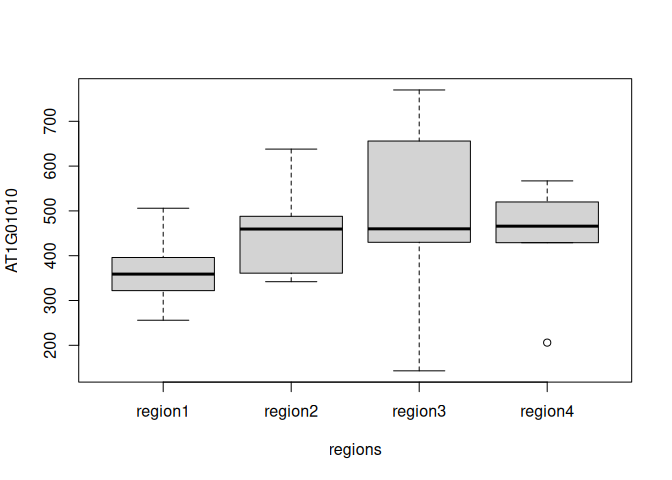
Boxplot for a single gene, separated by regions:
tc3 = data.frame(AT1G01010=tc$AT1G01010, regions=regions)
rownames(tc3) = rownames(tc)
boxplot(AT1G01010 ~ regions, data=tc3)
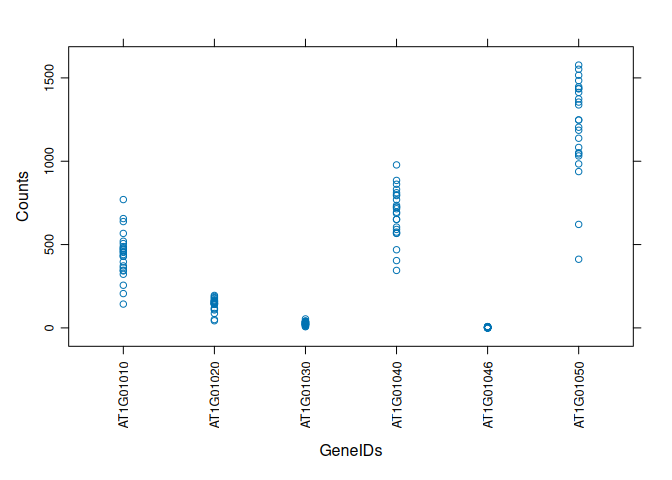
xyplot (bivariate scatterplot) of count data, separated by GeneID:
library(lattice)
xyplot(value ~ variable, data=tcm, scales=list(rot=90), xlab="GeneIDs", ylab="Counts")
Same xyplot, but also separated by regions:
xyplot(value ~ variable | regions, data=tcm, scales=list(rot=90), xlab="GeneIDs", ylab="Counts")
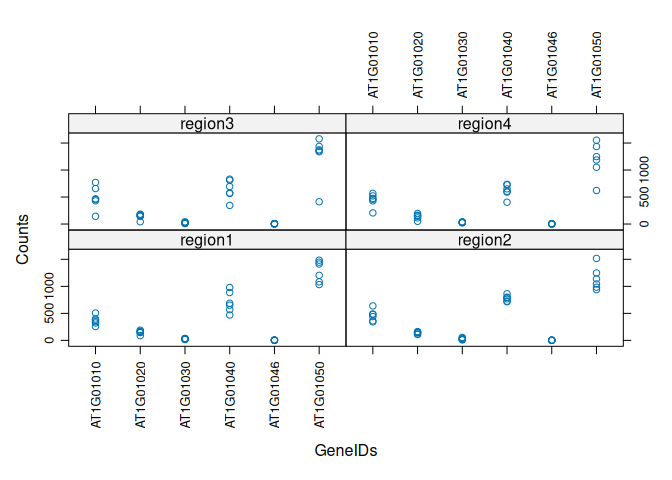
Same xyplot but with both GeneIDs and regions:
xyplot(value ~ variable + regions, data=tcm, scales=list(rot=90), xlab="GeneIDs", ylab="Counts")
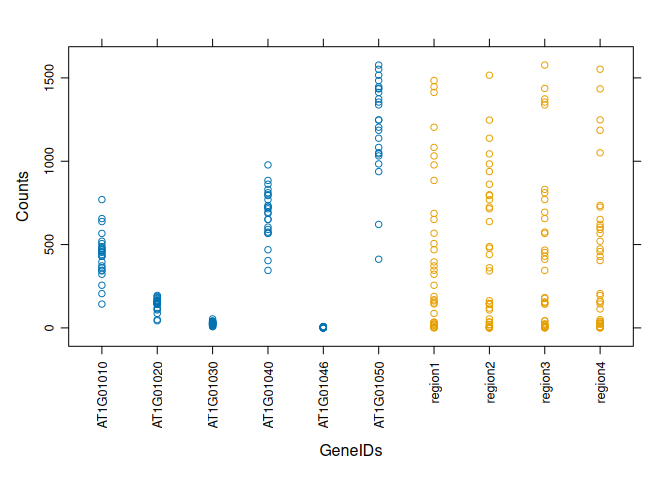
Boxplot for each GeneID in one plot:
boxplot(value ~ variable, data = tcm, las=2, ylab="Counts", xlab="")
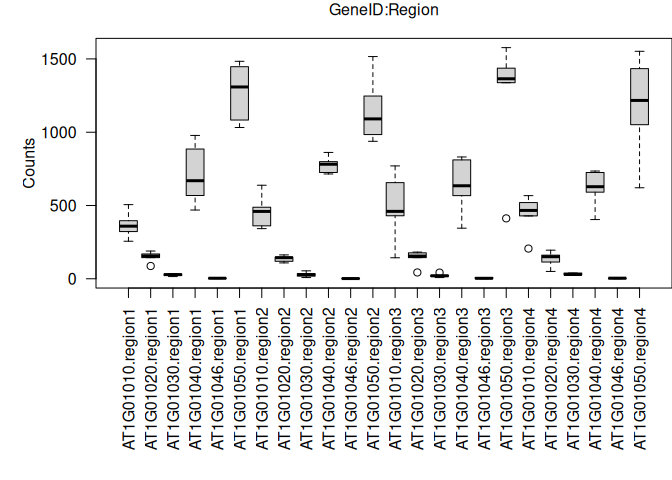
Now, the same boxplot but separating by GeneIDs and regions together:
# need to change the margins of the plot to fit the labels
par(mar = c(10, 5, 2, 0))
boxplot(value ~ variable * regions, data = tcm, las=2, ylab="Counts", xlab="")
# use mtext to create an x-axis label above the graph
mtext("GeneID:Region", side = 3, line = 1)
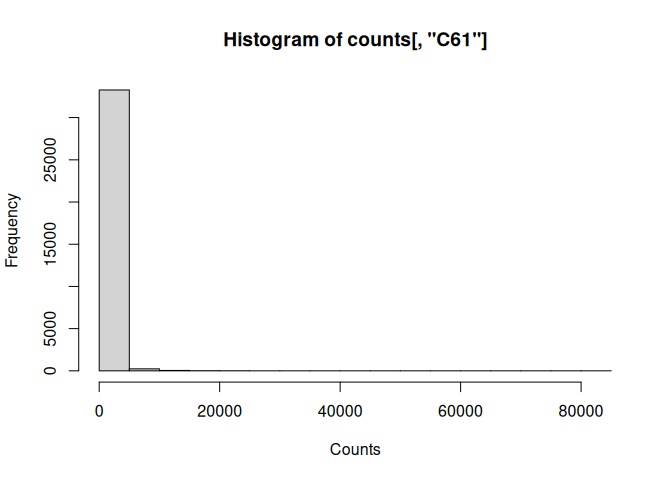
Simple histogram:
hist(counts[,"C61"], xlab="Counts")
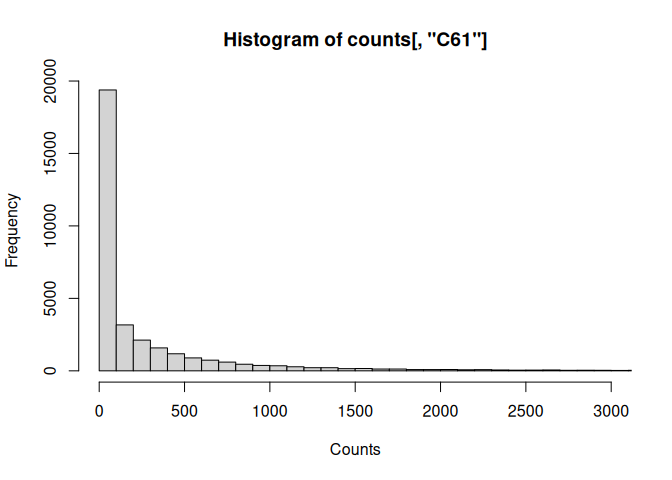
Refine the histogram:
hist(counts[,"C61"], xlab="Counts", breaks=1000, xlim=c(0,3000))
VISUALIZATION CHALLENGE
Download the gapminder dataset to your Rstudio session using the following URL:
Take a look at the dataset. Subset the data to only look at rows from 1982 . Then make a scatterplot of gdp vs life exp (using the plot function) of the subset, adding x and y labels. Find out how to log scale the x axis from the “plot.default” documentation.
Next, make a named vector of colors with the names being the continents. Use the vector to add colors to each point based on the continent for that point. You may need to look at the “col” section in the “plot.default” documentation. You can also use the “colors()” function to get a list of all of the available colors. Add a legend to the plot showing which colors correspond to which continent.
Finally, create a function that takes in a numeric vector and a minimum and maximum circle size. Within the function, take the square root of the vector, then use the min-max normalization method to normalize each element of the vector. Once you have normalized data, use those values to scale between the minimum and maximum circle size. Use this function to change the size of the circles to correspond to the population of the country using the “cex” parameter for the scatter plot.