
Bacterial RNASeq data reduction workflow
The challenge/difference
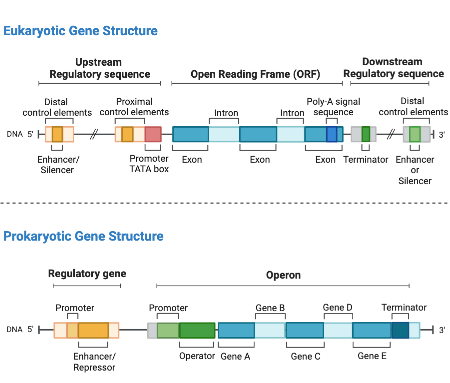
The major difference between RNASeq in prokaryotes and in eukaryotes is the gene structure. In generally, there are no introns in prokaryotic genes, therefore no splicing.
https://study.com/skill/practice/contrasting-the-regulation-of-gene-expression-in-prokaryotic-eukaryotic-organisms-questions.html
This intron-less structure in prokaryotic genes dictates how the RNASeq data should be processed, especially at the alignment stage.
-
One option is to use an alignment tool/aligner that allows for continuous mapping of the sequencing reads to the reference genome: Bowtie2. In the default mode (end-to-end mode), Bowtie2 searches for alignments that matches the sequencing reads from end to end. In another words, the alignments do not allow any trimming or clipping of bases from the sequencing reads. After the alignment, the quantification of gene expression can be done using featureCounts, by supplying the gene annotation information.
-
The other option is to use an aligner that aligns/pseudo-aligns the sequencing reads to the reference transcriptome: such as salmon.
-
An example of processing script for RNASeq data in prokaryotes. This assumes the sequencing data has gone through quality control: adapter trimming, quality trimming, length filtering,…
#!/bin/bash #SBATCH --nodes=1 #SBATCH --ntasks=8 #SBATCH --time=360 #SBATCH --mem=16000 # Memory pool for all cores (see also --mem-per-cpu) #SBATCH --partition=production #SBATCH --array=1-54 start=`date +%s` echo $HOSTNAME echo "My SLURM_ARRAY_TASK_ID: " $SLURM_ARRAY_TASK_ID sample=`sed "${SLURM_ARRAY_TASK_ID}q;d" samples.txt` annotation="References/gencode.vM26.annotation.gtf" outpath='02-Bowtie2' echo "SAMPLE: ${sample}" module load bowtie2 bowtie2 --very-sensitive -p 12 -x $refP/reference \ -1 R1.fastq -2 R2.fastq |\ samtools view -bh -@ 2 -m 5G -o $outpath/${sample}.bam - samtools index -b ${outpath}/${sample}.bam module load subread featureCounts -T 4 --verbose -s 1 \ -a ${annotation} \ -t gene \ -o ${outpath}//${sample}_featurecounts.txt \ ${outpath}/${sample}.bam \ > ${outpath}/${sample}_featurecounts.stdout \ 2> ${outpath}/${sample}_featurecounts.stderr end=`date +%s` runtime=$((end-start)) echo $runtime