
Introduction to Data
There are a few different data sets used throughout the workshop because no one data set meets the requirement for all aspects we are going to discuss. The first data set is a Xenium data set on Alzheimer’s disease model of mouse brain coronal section from one hemisphere. It is a data set provided by 10X Genomics on their website: https://www.10xgenomics.com/datasets/xenium-in-situ-analysis-of-alzheimers-disease-mouse-model-brain-coronal-sections-from-one-hemisphere-over-a-time-course-1-standard. The full data set includes 2 genotypes: wild type and Alzheimer’s disease model, and 3 time points per genotype. We are going to use only the time point at 5.7 months for the 2 genotypes in this workshop.
Explore the output from Xenium Onboard Analysis output
First, let’s create a project folder to keep all the workshop materials in. Then download the data and uncompress them and put them into their corresponding directories.
mkdir -p ~/Spatial_transcriptomics; cd Spatial_transcriptomics
mkdir Xenium_V1_FFPE_TgCRND8_5_7_months_outs; mv Xenium_V1_FFPE_TgCRND8_5_7_months_outs.zip Xenium_V1_FFPE_TgCRND8_5_7_months_outs/
mkdir Xenium_V1_FFPE_wildtype_5_7_months_outs; mv Xenium_V1_FFPE_wildtype_5_7_months_outs.zip Xenium_V1_FFPE_wildtype_5_7_months_outs/
Let’s take a look at the files inside Xenium_V1_FFPE_wildtype_5_7_months_outs folder as an example.
cd Xenium_V1_FFPE_wildtype_5_7_months_outs
ls
- analysis_summary.html - Contains summary metrics and automated secondary analysis results
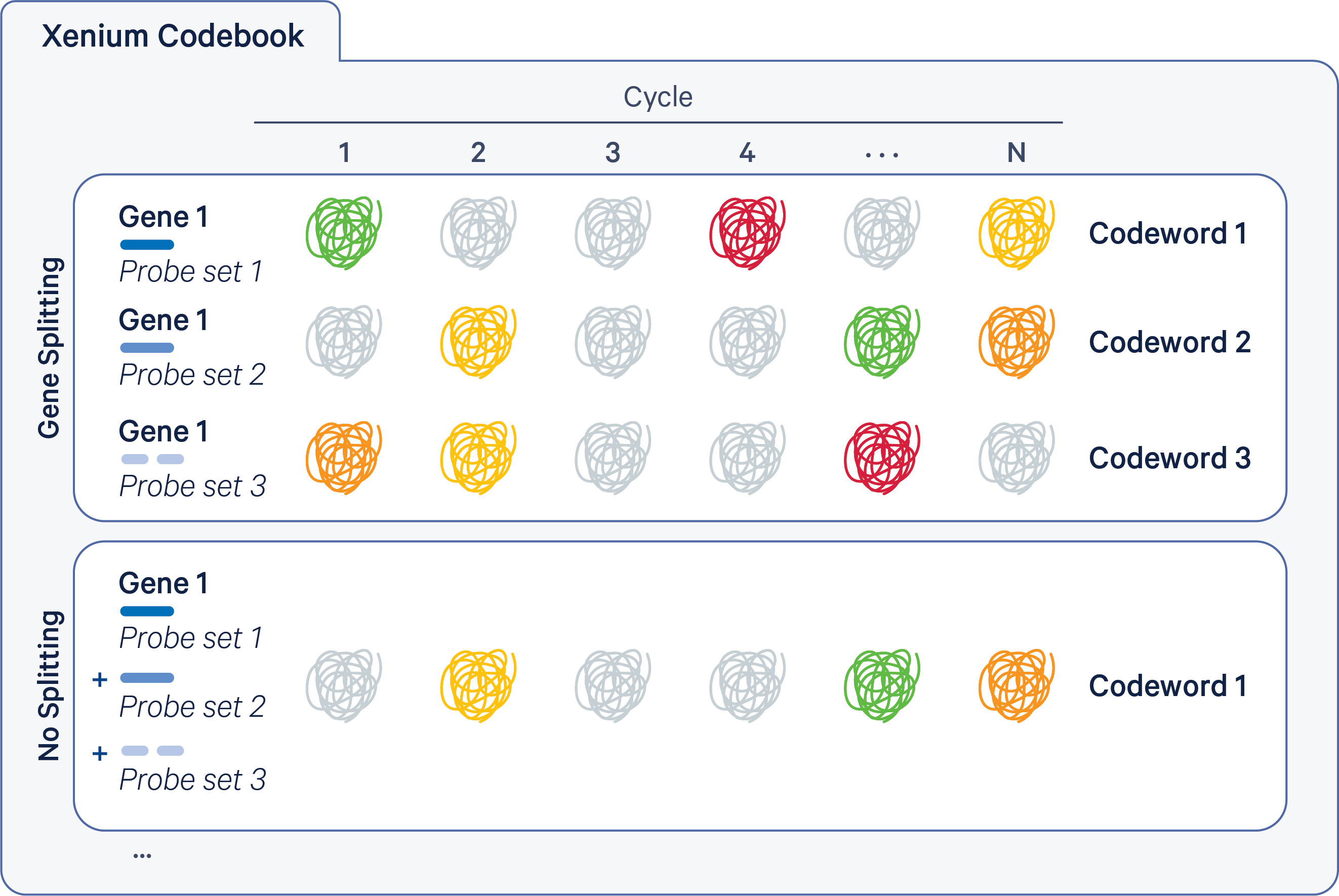
- gene_panel.json - transcript feature metadata
- morphology.ome.tif - 3D Z-stack of the DAPI image that can be useful to resegment cells, assess segmentation quality, and view data. Open with imageJ
- morphology_focus - available in Xenium experiment where 3 additional stain images available besides the nuclei DAPI stain image: boundary (ATP1A1/E-Cadherin/CD45) image, interior RNA (18S) image, and interior protein (alphaSMA/Vimentin) image
- cells.csv.gz - cell summary file with columns for:
- cell id
- X location of the cell centroid in microns
- Y location of the cell centroid in microns
- count of gene features for transcripts with Q-Score >= 20
- count of negative control probes
- count of genomic control codewords (Xenium Prime)
- count of negative control codewords
- count of unassigned codewords
- total counts of the probes and codewords
- 2D area covered by the cell in micron^2
- 2D area covered by the nucleus in micron^2
- count of detected nuclei
- cell segmentation method
- cells.parquet - cell summary in Parquet format that allows faster loading and reading of data
- cells.zarr.zip - cell segmentation file in Zarr format that contains segmentation masks and boundaries for nuclei and cells. It’s used to assign transcripts to cells
- nucleus_boundaries.csv.gz and cell_boundaries.csv.gz are nucleus and cell boundaries in csv format. Each row represents a vertex in the boundary polygon of the cell/nucleus and the boundaries points for each cell/nucleus are listed in clockwise order.
- transcripts.parquet contains data to evaluate transcript quality and localization.
- transcript id
- cell id
- binary value to indicate if the transcript falls within the segmented nuccleus of a cell
- gene or control name
- X location of the transcript in micron
- Y location of the transcript in micron
- Z location of the transcript in micron
- phred-scaled quality value (Q-Score) estimating the probability of incorrect call
- FOV name
- the distance between the transcript and the nearest nucleus boundary in micron based on segmentation mask boundaries.
- an integer index for each codeword used to decode transcripts
- codeword category
- value to indicate whether transcript feature is “Gene Expression” or not
- cell_feature_matrix - cell-feature matrix output from XOA in three formats: a standard text-based format (the Market Exchange Format (MEX)), the Hierarchical Data Format (HDF5), and the Zarr format. They only include transcripts that pass the default quality value threshold of Q20 and are assigned to cells.
- analysis folder contains outputs from XOA pipeline: clustering, differential expression, principal component analysis and UMAP.
- aux_outputs
The output from CosMx AtoMx pipeline
Bruker AtoMx interface provides export functions to download necessary files to be used with community developed tools. One may export flat files (text based, csv).
- count matrix
- cell metadata
- transcripts
- polygons
- FOV positions
Tertiary analysis objects, such as Seurat object and TileDB array, can be exported as well. User has the option to include transcript coordinates and polygon coordinates in the exported Seurat object. But these produce large data files. Morphology 2D data may be exported as well, but produces very large data.
The output from Visium HD platform
10X Genomics Visium HD runs generate fastq files. The first step in analysis is to run Space Ranger to translate the raw sequencing data to location decoded gene expression matrix. The input files required for Space Ranger.
- fastq files
- reference file
- probe set file
- slide layout file
- CytAssist-captured image
- optionally, a high resolution microscope image in either brightfield and fluorescence image can be added