
Using the Iso-Seq Application on SMRTlink and BioConda
Elizabeth Tseng, Principal Scientist, PacBio
Why use Iso-Seq analysis?
ISO-SEQ ANALYSIS MAIN FEATURES
- No reference genome required
- No transcriptome assembly required
- Recovers full-length (5’ to 3’) transcripts
- Yields highly accurate (>99%) transcripts
HIFI READS FROM CCS
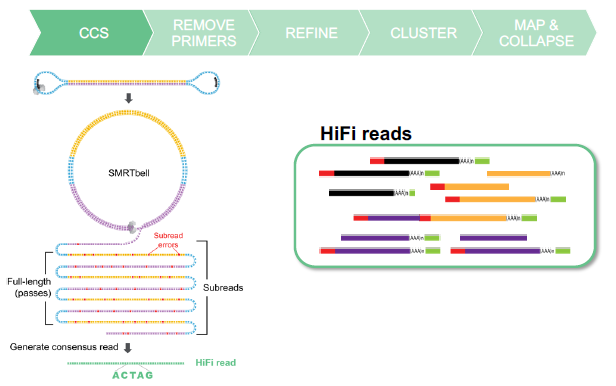
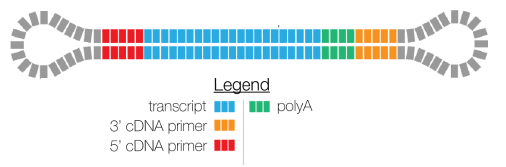
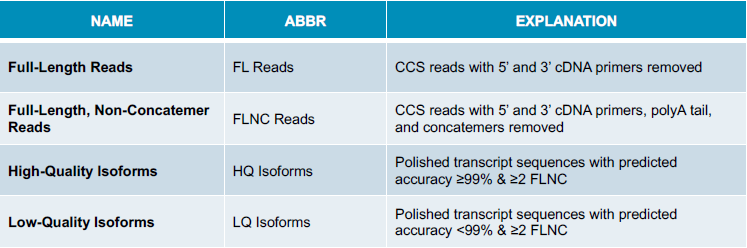
FULL-LENGTH READS HAVE 5’ AND 3’ PRIMERS
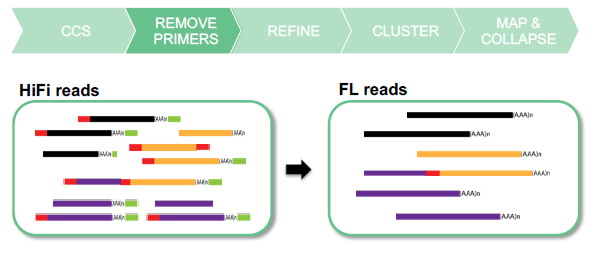
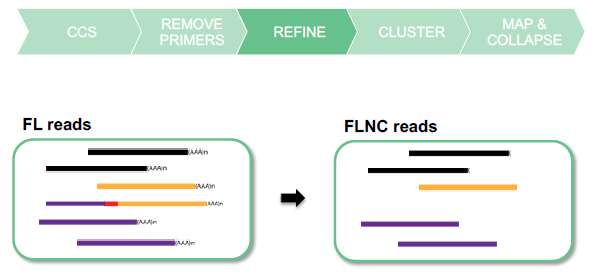
REMOVE CONCATEMERS AND POLY(A) TAILS
CLUSTER TO GET ISOFORMS
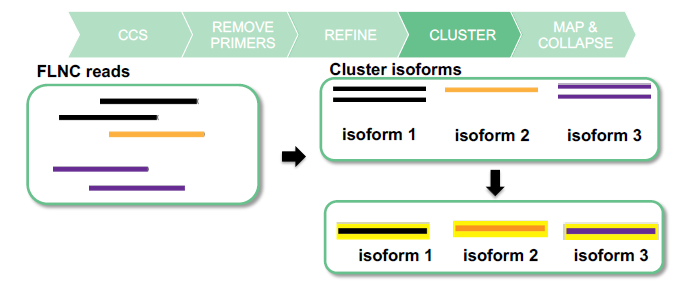
- High Quality (HQ): accuracy ≥99% AND ≥2 FLNC read support
- Low Quality (LQ): accuracy <99% OR <2 FLNC read support
Note: The pdf is wrong. Low quality is anything that is lower than 99% or has less than 2 FLNC read support.
MAP AND COLLAPSE ISOFORMS
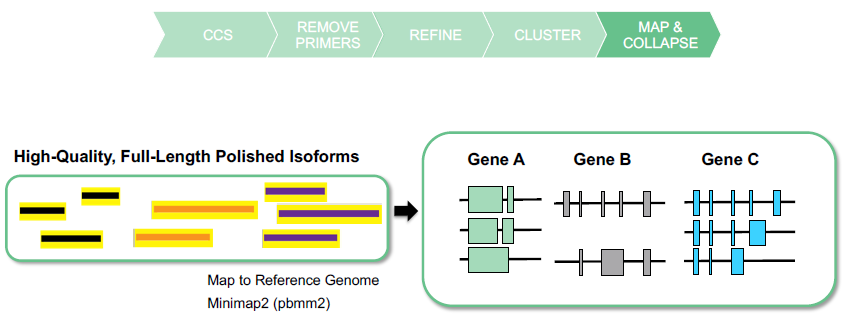
BENEFITS OF ISO-SEQ ANALYSIS APPLICATION
- High-quality transcripts
- Full-Length Non-concatemer reads
- Mapped & collapsed isoforms
- Removes artifacts
- Removes poly(A) tails
Iso-Seq Analysis Using pbBioConda
INSTRUCTIONS TUTORIAL
Follow the instructions tutorial for installing all the software needed.
- If you do not have an HPC server to install pbbioconda, you should have already:
- Create an AWS account
- Create an AWS Linux Instance to run Iso-Seq 3 Analysis Pipeline
- Connect to your AWS Instance
- Upgrades and Install Software
DOWNLOAD THE DATA here (we will be using the data to do practice questions in the tutorial)
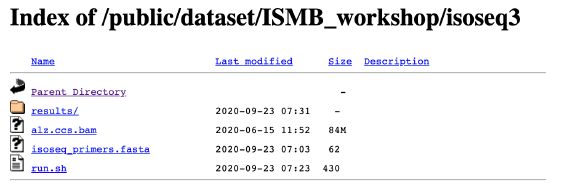
Example:
$ wget –nv https://downloads.pacbcloud.com/public/dataset/ISMB_workshop/isoseq3/alz.ccs.bam
SPECIFY ISO-SEQ PRIMERS
$ more primers.fasta
INPUT CCS BAM FILE
$ samtools view -h alz.ccs.bam
REFERENCE GENOME
$ grep '>’ hg38.fa # to list the headers per chromosome
SOFTWARE INSTALLATION CHECK
Access to your conda environment
$ source activate <name of your environment>
Check your installation
$ isoseq3 --version
$ lima --version
$ pbmm2 –-version
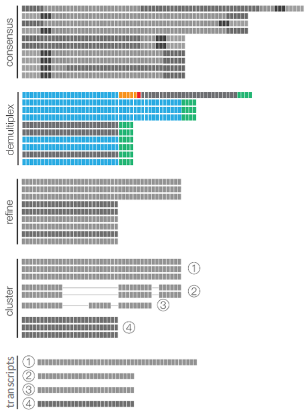
ISO-SEQ WORKFLOW
Click here if you want to look at the entire Iso Seq workflow on PacBio’s GitHub page
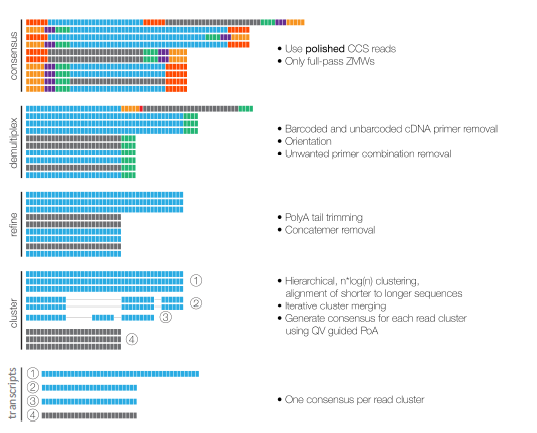
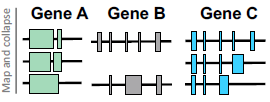
- Align to reference genome
- Remove redundancy
PRIMER REMOVAL & DEMULTIPLEXING
Command line:
lima --isoseq --dump-clips --peek-guess -j 24\
Input files:
- alz.ccs.bam #HiFi reads
- isoseq_primers.fasta #Iso-Seq primers
Output files:
- alz.demult.bam
Options:
- –isoseq #specialized isoseq option for lima
- –dump-clips # show the clipped primers
- –peek-guess # remove spurious false positive signal
- -j 24 # Number of threads to use
After completion, you will see the following files:
$ ls -ltrh

TRIMMING POLY(A) TAILS AND CONCATEMER REMOVAL

Command line:
isoseq3 refine --require-polya\
Input files:
- alz.demult.5p–3p.bam
- isoseq_primers.fasta
Output files:
- alz.flnc.bam
Options:
- –require-polya #if your transcripts have a polyA tail
After completion, you will see the following files:
$ ls -ltrh

ISOFORMS

Command line:
isoseq3 cluster alz.flnc.bam alz.polished.bam \
--verbose --use-qvs
Input files:
- alz.flnc.bam
Output files:
- alz.flnc.bam
Options:
- –verbose #if your transcripts have a polyA tail
- –use-qvs #Use CCS QVs, sets –poa-cov 100
After completion, you will see the following files:
$ ls -ltrh
 Note: Because the ccs input is Polished, the isoseq3 cluster output is already polished!
Note: Because the ccs input is Polished, the isoseq3 cluster output is already polished!
MAP
Command line:
pbmm2 align hg38.fa alz.polished.hq.bam alz.aligned.bam
-j 24 --preset ISOSEQ –-sort --log-level INFO
Note: The pdf is wrong. It should be –sort, not -sort.
Input files:
- alz.polished.hq.bam
- hg38.fa
Output files:
- alz.aligned.bam
Options:
- -j 24 #Number of threads to use
- –preset ISOSEQ #select the alignment mode
- –sort #Generate sorted BAM file
- –log-level INFO #show progress
After completion, you will see the following files:
$ ls –ltrh
COLLAPSE
Command line:
isoseq3 collapse alz.aligned.bam alz.collapsed.gff
Input files:
- alz.aligned.bam
Output files:
- alz.collapsed.gff
After completion, you will see the following files:
$ ls –ltrh

PUBLICLY AVAILABLE ISO-SEQ DATA SETS here if you’re interested
ISO-SEQ ANALYSIS TERMINOLOGY
The pdf to this documentation can be found here
Iso-Seq pipeline with Bioconda and visualization using UCSC and IGV & Cupcake (hands on)
The Iso Seq Bioinformatics Tutorial we are using is here. See ‘1. isoseq3’ and answer the corresponding practice questions!
Note: We did not have time to do Cupcake during the workshop so try it out yourself and if you have questions feel free to reach out! See ‘2. Cupcake Fun’ and answer the corresponding practice questions!