
Reading in the experiment data and exploring the Phyloseq object
The phyloseq package has become a convenient way a managing micobial community data, filtering and visualizing data, performing analysis such as ordination. Along with the standard R environment and packages vegan and vegetarian you can perform virtually any analysis. Today we will
- Load phyloseq object
- Perform some QA/QC
- Filter Data
- Graphical Summaries
- Ordination
- Differential Abundances
Load our libraries
# Set up global options for nice reports and keeping figures:
knitr::opts_chunk$set(fig.width=14, fig.height=8, fig.align="center",
warning=FALSE, message=FALSE)
Lets start by loading libraries
library(phyloseq)
library(phangorn)
library(ggplot2)
nice_colors = c("#999999", "#E69F00", "#56B4E9","#e98756","#c08160","#5800e6", "#CDDC49", "#C475D3",
"#E94B30", "#233F57", "#FEE659", "#A1CFDD", "#F4755E", "#D6F6F7","#EB6D58", "#6898BF")
Read in the dataset,
Lets first read in the Phyloseq object, this is the full dataset 197 generated from the same pipeline used in the data reduction section.
load(file="phyloseq_nochim_silva.RData")
ls()
## [1] "nice_colors" "ps.silva.nochim"
# set the data set used for analysis
ps = ps.silva.nochim
head(otu_table(ps))[,1:10]
## OTU Table: [10 taxa and 6 samples]
## taxa are columns
## ASV1 ASV2 ASV3 ASV4 ASV5 ASV6 ASV7 ASV8 ASV9 ASV10
## Bs1_10C_A0 2041 46 944 234 506 119 381 84 150 1140
## Bs1_10C_A5 965 45 961 163 609 304 165 110 256 576
## Bs1_10C_B0 1151 10 731 184 387 73 192 58 90 429
## Bs1_10C_B5 525 21 586 155 361 143 129 39 149 550
## Bs1_10C_C0 1218 10 560 165 320 85 327 52 115 472
## Bs1_10C_C5 352 15 294 61 213 108 68 39 95 242
head(sample_data(ps))
## SampleID Group Temp Replicate
## Bs1_10C_A0 Bs1_10C_A0 Bs1 10C A0
## Bs1_10C_A5 Bs1_10C_A5 Bs1 10C A5
## Bs1_10C_B0 Bs1_10C_B0 Bs1 10C B0
## Bs1_10C_B5 Bs1_10C_B5 Bs1 10C B5
## Bs1_10C_C0 Bs1_10C_C0 Bs1 10C C0
## Bs1_10C_C5 Bs1_10C_C5 Bs1 10C C5
head(tax_table(ps))
## Taxonomy Table: [6 taxa by 7 taxonomic ranks]:
## Kingdom Phylum Class Order
## ASV1 "Bacteria" "Desulfobacterota" "Syntrophobacteria" "Syntrophobacterales"
## ASV2 "Bacteria" "Cyanobacteria" "Cyanobacteriia" "Chloroplast"
## ASV3 "Bacteria" "Firmicutes" "Bacilli" "Bacillales"
## ASV4 "Bacteria" "Myxococcota" "bacteriap25" NA
## ASV5 "Bacteria" "Firmicutes" "Bacilli" "Bacillales"
## ASV6 "Bacteria" "Firmicutes" "Clostridia" "Clostridiales"
## Family Genus Species
## ASV1 "Syntrophobacteraceae" "Syntrophobacter" NA
## ASV2 NA NA NA
## ASV3 "Planococcaceae" "Paenisporosarcina" NA
## ASV4 NA NA NA
## ASV5 "Planococcaceae" "Paenisporosarcina" NA
## ASV6 "Clostridiaceae" "Clostridium sensu stricto 1" NA
head(refseq(ps))
## DNAStringSet object of length 6:
## width seq names
## [1] 255 TACGGAGGGTGCGAGCGTTATTC...CGAAAGCGTGGGGAGCAAACAGG ASV1
## [2] 253 GACGGAGGATGCAAGTGTTATCC...CGAAAGCTAGGGTAGCAAATGGG ASV2
## [3] 253 TACGTAGGTGGCAAGCGTTGTCC...CGAAAGCGTGGGGAGCAAACAGG ASV3
## [4] 253 TACAGAGGGTGCAAGCGTTGTTC...CGAAAGCGTGGGGAGCAAACAGG ASV4
## [5] 253 TACGTAGGTGGCAAGCGTTGTCC...CGAAAGCGTGGGGAGCAAACAGG ASV5
## [6] 253 TACGTAGGTGGCAAGCGTTGTCC...CGAAAGCGTGGGGAGCAAACAGG ASV6
head(rank_names(ps))
## [1] "Kingdom" "Phylum" "Class" "Order" "Family" "Genus"
sample_variables(ps)
## [1] "SampleID" "Group" "Temp" "Replicate"
Root the phylogenetic tree
Some analysis require the phylogenetic tree to be rooted. We use phanghorn root command to set our root.
set.seed(1)
is.rooted(phy_tree(ps))
## [1] FALSE
phy_tree(ps) <- root(phy_tree(ps), sample(taxa_names(ps), 1), resolve.root = TRUE)
is.rooted(phy_tree(ps))
## [1] TRUE
The Phyloseq Object
A lot of information is in this object, spend somem time to get to know it.
- Explore the sample data, how many Groups do we have? Temps? patterns of replicate?
- Using the
@operator to access class objects are there any other info in the ps object not already looked at/ -
- Use
?"phyloseq-class"to get information on the class and object.
- Use
ps
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1500 taxa and 197 samples ]
## sample_data() Sample Data: [ 197 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 1500 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1500 tips and 1499 internal nodes ]
## refseq() DNAStringSet: [ 1500 reference sequences ]
Drawing our first plot
ps
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1500 taxa and 197 samples ]
## sample_data() Sample Data: [ 197 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 1500 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1500 tips and 1499 internal nodes ]
## refseq() DNAStringSet: [ 1500 reference sequences ]
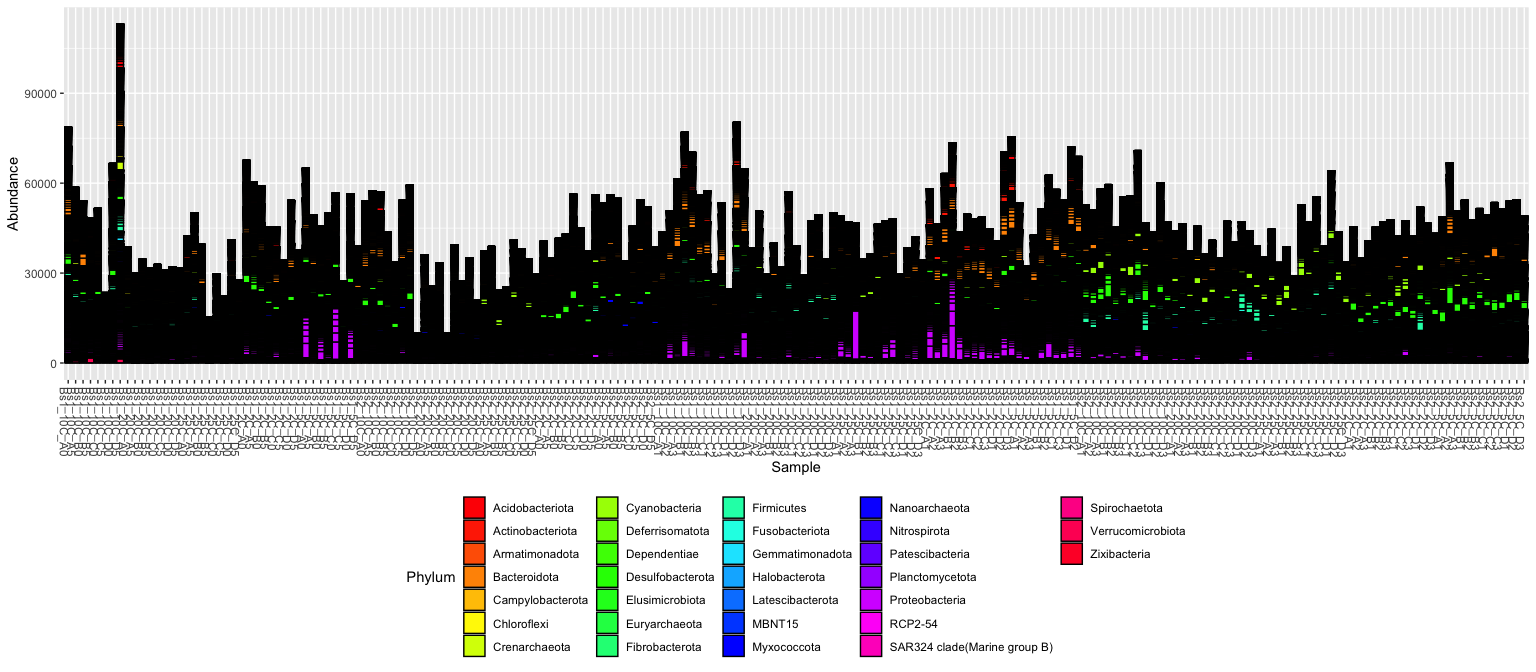
plot_bar(ps, fill = "Phylum") + theme(legend.position="bottom" ) + scale_fill_manual(values = rainbow(length(unique(tax_table(ps)[,"Phylum"]))))
Cleanup
Save object
dir.create("rdata_objects", showWarnings = FALSE)
save(ps, file=file.path("rdata_objects", "initial_rooted.Rdata"))
Get next Rmd
download.file("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2021-May-Microbial-Community-Analysis/master/data_analysis/mca_part2.Rmd", "mca_part2.Rmd")
Record session information
sessionInfo()
## R version 4.0.3 (2020-10-10)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] ggplot2_3.3.3 phangorn_2.7.0 ape_5.5 phyloseq_1.34.0
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.6 lattice_0.20-44 prettyunits_1.1.1
## [4] Biostrings_2.58.0 assertthat_0.2.1 digest_0.6.27
## [7] foreach_1.5.1 utf8_1.2.1 R6_2.5.0
## [10] plyr_1.8.6 stats4_4.0.3 evaluate_0.14
## [13] highr_0.9 pillar_1.6.1 zlibbioc_1.36.0
## [16] rlang_0.4.11 progress_1.2.2 data.table_1.14.0
## [19] vegan_2.5-7 jquerylib_0.1.4 S4Vectors_0.28.1
## [22] Matrix_1.3-3 rmarkdown_2.8 labeling_0.4.2
## [25] splines_4.0.3 stringr_1.4.0 igraph_1.2.6
## [28] munsell_0.5.0 compiler_4.0.3 xfun_0.23
## [31] pkgconfig_2.0.3 BiocGenerics_0.36.1 multtest_2.46.0
## [34] mgcv_1.8-35 htmltools_0.5.1.1 biomformat_1.18.0
## [37] tidyselect_1.1.1 tibble_3.1.2 quadprog_1.5-8
## [40] IRanges_2.24.1 codetools_0.2-18 permute_0.9-5
## [43] fansi_0.4.2 withr_2.4.2 crayon_1.4.1
## [46] dplyr_1.0.6 MASS_7.3-54 rhdf5filters_1.2.1
## [49] grid_4.0.3 nlme_3.1-152 jsonlite_1.7.2
## [52] gtable_0.3.0 lifecycle_1.0.0 DBI_1.1.1
## [55] magrittr_2.0.1 scales_1.1.1 stringi_1.6.2
## [58] farver_2.1.0 XVector_0.30.0 reshape2_1.4.4
## [61] bslib_0.2.5.1 ellipsis_0.3.2 vctrs_0.3.8
## [64] generics_0.1.0 fastmatch_1.1-0 Rhdf5lib_1.12.1
## [67] iterators_1.0.13 tools_4.0.3 ade4_1.7-16
## [70] Biobase_2.50.0 glue_1.4.2 purrr_0.3.4
## [73] hms_1.1.0 parallel_4.0.3 survival_3.2-11
## [76] yaml_2.2.1 colorspace_2.0-1 rhdf5_2.34.0
## [79] cluster_2.1.2 knitr_1.33 sass_0.4.0