
Filtering taxa and samples
Load our libraries
# Set up global options for nice reports and keeping figures:
knitr::opts_chunk$set(fig.width=14, fig.height=8, fig.align="center",
warning=FALSE, message=FALSE)
Lets start by loading libraries
library(phyloseq)
library(phangorn)
library(ggplot2)
library(gridExtra)
nice_colors = c("#999999", "#E69F00", "#56B4E9","#e98756","#c08160","#5800e6", "#CDDC49", "#C475D3",
"#E94B30", "#233F57", "#FEE659", "#A1CFDD", "#F4755E", "#D6F6F7","#EB6D58", "#6898BF")
Load prior results
load(file=file.path("rdata_objects", "initial_rooted.Rdata"))
Now lets filter out samples (outliers and low performing samples)
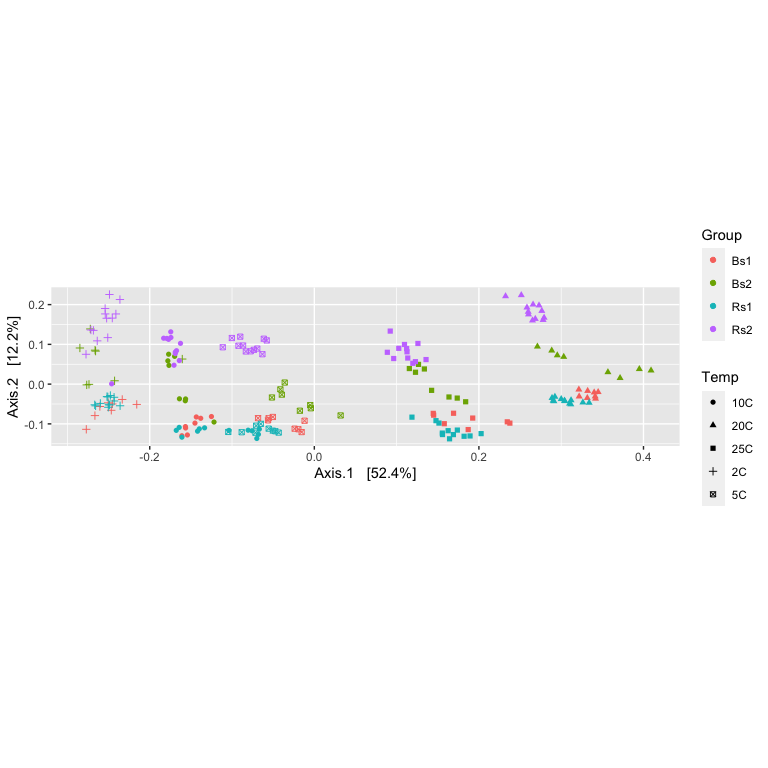
Some simple ordination looking for ‘outlier’ samples, first we variance stabilize the data with a log transform, the perform MDS using bray’s distances
logt = transform_sample_counts(ps, function(x) log(1 + x) )
out.mds.logt <- ordinate(logt, method = "MDS", distance = "bray")
evals <- out.mds.logt$values$Eigenvalues
plot_ordination(logt, out.mds.logt, type = "samples",
color = "Group", shape = "Temp") + labs(col = "Group") +
coord_fixed(sqrt(evals[2] / evals[1]))
Show taxa proportions per sample (quickplot)
grid.arrange(nrow = 2,
qplot(as(otu_table(logt),"matrix")[which.min(sample_sums(logt)),], geom = "histogram", bins=50) +
xlab("Relative abundance"),
qplot(as(otu_table(logt),"matrix")[which.max(sample_sums(logt)),], geom = "histogram", bins=50) +
xlab("Relative abundance")
)
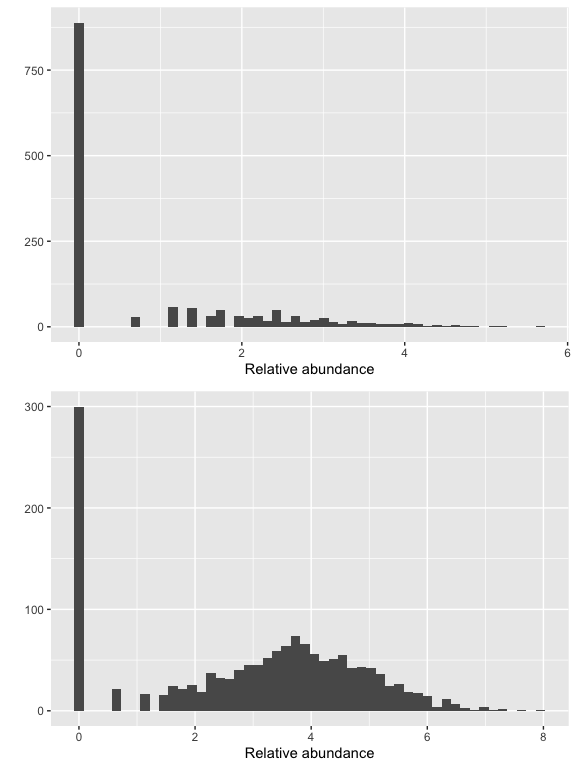
Nothing so far seems to stand out and our read counts ASV look good from our prior QA, so we won’t remove any samples in this experiment.
However if we wanted to prune samples, say removing all <10K reads, the code below would do so.
ps.pruned <- prune_samples(sample_sums(ps)>=10000, ps)
ps.pruned
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1500 taxa and 195 samples ]
## sample_data() Sample Data: [ 195 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 1500 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1500 tips and 1499 internal nodes ]
## refseq() DNAStringSet: [ 1500 reference sequences ]
- So how many samples were pruned?
Taxa Filtering
Whole phylum filtering
Now lets investigate low prevelance/abundance phylum and subset them out.
Lets generate a prevelance table (number of samples each taxa occurs in) for each taxa.
prevalenceDF = data.frame(Prevalence = colSums(otu_table(ps) > 0 ),
TotalAbundance = colSums(otu_table(ps)),
tax_table(ps)
)
idxAbundance = order(prevalenceDF$TotalAbundance, decreasing=T)
idxPrevalence = order(prevalenceDF$Prevalence, decreasing=T)
summary_prevalence <- plyr::ddply(prevalenceDF, "Phylum", function(df1){
data.frame(mean_prevalence=mean(df1$Prevalence),totalAbundance=sum(df1$TotalAbundance,na.rm = T),stringsAsFactors = F)
})
summary_prevalence[order(summary_prevalence$totalAbundance, decreasing = FALSE),]
## Phylum mean_prevalence totalAbundance
## 10 Dependentiae 114.0000 2071
## 12 Elusimicrobiota 146.0000 2239
## 31 Zixibacteria 131.0000 2610
## 28 SAR324 clade(Marine group B) 132.0000 2703
## 16 Fusobacteriota 59.0000 5677
## 24 Patescibacteria 92.0000 6053
## 9 Deferrisomatota 148.5000 6170
## 3 Armatimonadota 119.6667 8416
## 18 Halobacterota 105.0000 10292
## 13 Euryarchaeota 122.6667 10738
## 19 Latescibacterota 128.7500 14570
## 29 Spirochaetota 116.3750 21799
## 14 Fibrobacterota 165.5000 25583
## 27 RCP2-54 111.0000 27067
## 5 Campylobacterota 118.8000 33503
## 22 Nanoarchaeota 125.7273 39722
## 25 Planctomycetota 140.8571 42482
## 20 MBNT15 132.0000 43641
## 7 Crenarchaeota 137.7857 113557
## 23 Nitrospirota 137.0417 125623
## 17 Gemmatimonadota 125.9231 178671
## 21 Myxococcota 125.6809 244296
## 30 Verrucomicrobiota 136.2459 247497
## 8 Cyanobacteria 137.6667 331544
## 2 Actinobacteriota 150.0388 629465
## 6 Chloroflexi 138.4236 684725
## 1 Acidobacteriota 133.2391 692709
## 15 Firmicutes 153.3011 720766
## 11 Desulfobacterota 147.0940 938085
## 4 Bacteroidota 123.6171 1459098
## 26 Proteobacteria 129.4045 2409594
Using the table above, determine the phyla to filter, filtering 0.1% experiment wide abundance.
sum(summary_prevalence$totalAbundance)*0.001
## [1] 9080.966
table(summary_prevalence$totalAbundance/sum(summary_prevalence$totalAbundance) >= 0.001)
##
## FALSE TRUE
## 8 23
keepPhyla <- summary_prevalence$Phylum[summary_prevalence$totalAbundance/sum(summary_prevalence$totalAbundance) >= 0.001]
ps.1 = subset_taxa(ps, Phylum %in% keepPhyla)
ps.1
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1487 taxa and 197 samples ]
## sample_data() Sample Data: [ 197 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 1487 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1487 tips and 1486 internal nodes ]
## refseq() DNAStringSet: [ 1487 reference sequences ]
summary_prevalence <- summary_prevalence[summary_prevalence$Phylum %in% keepPhyla,]
summary_prevalence[order(summary_prevalence$totalAbundance, decreasing = FALSE),]
## Phylum mean_prevalence totalAbundance
## 18 Halobacterota 105.0000 10292
## 13 Euryarchaeota 122.6667 10738
## 19 Latescibacterota 128.7500 14570
## 29 Spirochaetota 116.3750 21799
## 14 Fibrobacterota 165.5000 25583
## 27 RCP2-54 111.0000 27067
## 5 Campylobacterota 118.8000 33503
## 22 Nanoarchaeota 125.7273 39722
## 25 Planctomycetota 140.8571 42482
## 20 MBNT15 132.0000 43641
## 7 Crenarchaeota 137.7857 113557
## 23 Nitrospirota 137.0417 125623
## 17 Gemmatimonadota 125.9231 178671
## 21 Myxococcota 125.6809 244296
## 30 Verrucomicrobiota 136.2459 247497
## 8 Cyanobacteria 137.6667 331544
## 2 Actinobacteriota 150.0388 629465
## 6 Chloroflexi 138.4236 684725
## 1 Acidobacteriota 133.2391 692709
## 15 Firmicutes 153.3011 720766
## 11 Desulfobacterota 147.0940 938085
## 4 Bacteroidota 123.6171 1459098
## 26 Proteobacteria 129.4045 2409594
Individual Taxa Filtering
Subset to the remaining phyla by prevelance.
prevalenceDF1 = subset(prevalenceDF, Phylum %in% get_taxa_unique(ps.1, taxonomic.rank = "Phylum"))
ggplot(prevalenceDF1, aes(TotalAbundance,Prevalence / nsamples(ps.1),color=Phylum)) +
# Include a guess for filtering intercept
geom_hline(yintercept = 0.25, alpha = 0.5, linetype = 2) + geom_point(size = 2, alpha = 0.7) +
scale_x_log10() + xlab("Total Abundance") + ylab("Prevalence [Frac. Samples]") +
facet_wrap(~Phylum) + theme(legend.position="none")
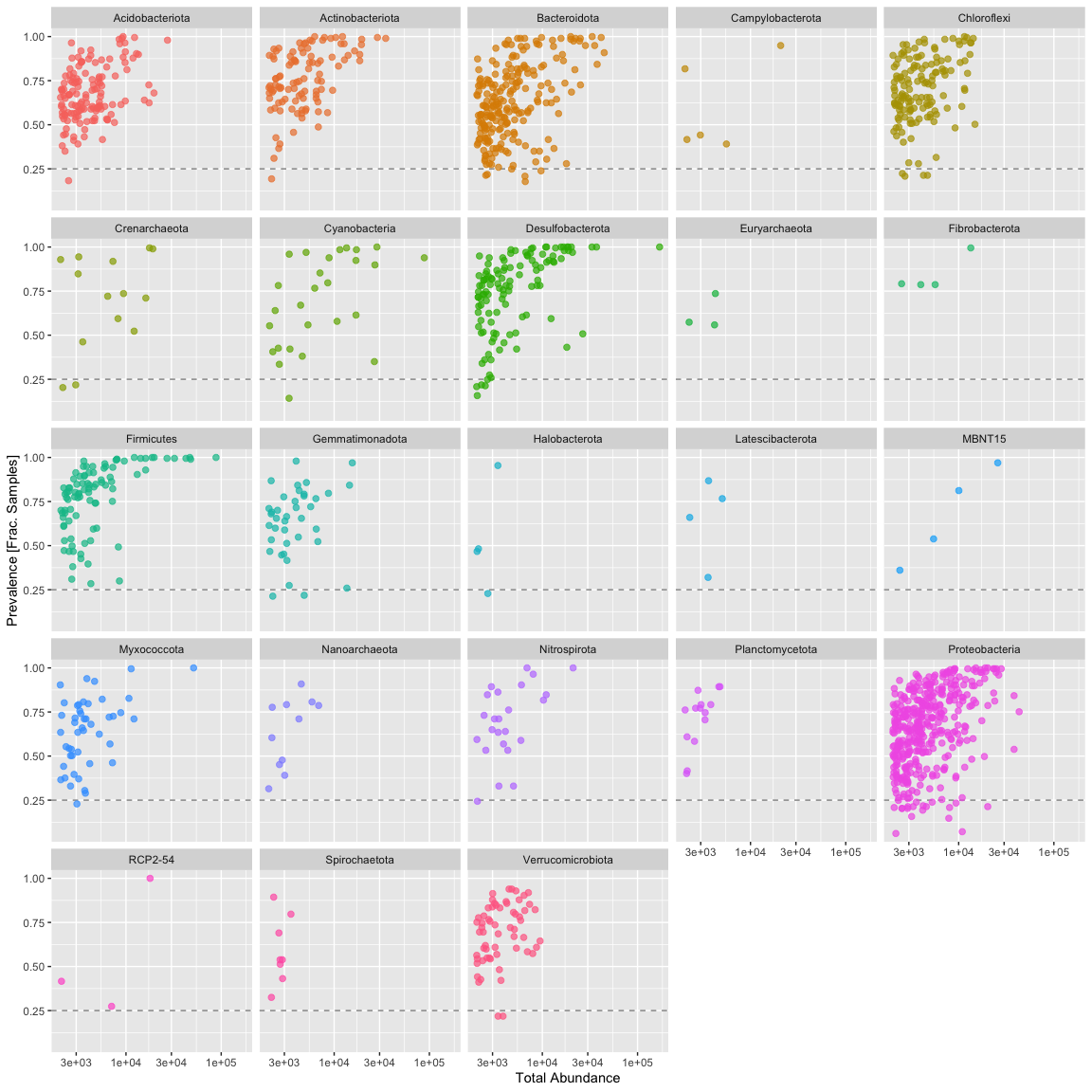 Sometimes you see a clear break, however we aren’t seeing one here. In this case I’m mostly interested in those organisms consistently present in the dataset, so I’m removing all taxa present in less than 25% of samples.
Sometimes you see a clear break, however we aren’t seeing one here. In this case I’m mostly interested in those organisms consistently present in the dataset, so I’m removing all taxa present in less than 25% of samples.
# Define prevalence threshold as 10% of total samples ~ set of replicates
prevalenceThreshold = 0.25 * nsamples(ps.1)
prevalenceThreshold
## [1] 49.25
# Execute prevalence filter, using `prune_taxa()` function
keepTaxa = rownames(prevalenceDF1)[(prevalenceDF1$Prevalence >= prevalenceThreshold)]
length(keepTaxa)
## [1] 1441
ps.1 = prune_taxa(keepTaxa, ps.1)
ps.1
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 1441 taxa and 197 samples ]
## sample_data() Sample Data: [ 197 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 1441 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 1441 tips and 1440 internal nodes ]
## refseq() DNAStringSet: [ 1441 reference sequences ]
Agglomerate taxa at the Genus level (combine all with the same name) removing all asv without genus level assignment.
length(get_taxa_unique(ps.1, taxonomic.rank = "Family"))
## [1] 172
ps.1 = tax_glom(ps.1, "Family", NArm = TRUE)
ps.1 = tax_glom(ps.1, "Genus", NArm = FALSE)
ps.1
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 171 taxa and 197 samples ]
## sample_data() Sample Data: [ 197 samples by 4 sample variables ]
## tax_table() Taxonomy Table: [ 171 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 171 tips and 170 internal nodes ]
## refseq() DNAStringSet: [ 171 reference sequences ]
## out of curiosity how many "reads" does this leave us at???
sum(colSums(otu_table(ps.1)))
## [1] 7377131
- So what percentage is that? from the original dataset?
Cleanup
Save object
dir.create("rdata_objects", showWarnings = FALSE)
save(list=c("ps","ps.1"), file=file.path("rdata_objects", "filtered_phyloseq.RData"))
Get next Rmd
download.file("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2021-May-Microbial-Community-Analysis/master/data_analysis/mca_part4.Rmd", "mca_part4.Rmd")
Record session information
sessionInfo()
## R version 4.0.3 (2020-10-10)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Big Sur 10.16
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] gridExtra_2.3 ggplot2_3.3.3 phangorn_2.7.0 ape_5.5
## [5] phyloseq_1.34.0
##
## loaded via a namespace (and not attached):
## [1] Biobase_2.50.0 sass_0.4.0 jsonlite_1.7.2
## [4] splines_4.0.3 foreach_1.5.1 bslib_0.2.5.1
## [7] assertthat_0.2.1 highr_0.9 stats4_4.0.3
## [10] yaml_2.2.1 progress_1.2.2 pillar_1.6.1
## [13] lattice_0.20-44 glue_1.4.2 quadprog_1.5-8
## [16] digest_0.6.27 XVector_0.30.0 colorspace_2.0-1
## [19] htmltools_0.5.1.1 Matrix_1.3-3 plyr_1.8.6
## [22] pkgconfig_2.0.3 zlibbioc_1.36.0 purrr_0.3.4
## [25] scales_1.1.1 tibble_3.1.2 mgcv_1.8-35
## [28] generics_0.1.0 farver_2.1.0 IRanges_2.24.1
## [31] ellipsis_0.3.2 withr_2.4.2 BiocGenerics_0.36.1
## [34] survival_3.2-11 magrittr_2.0.1 crayon_1.4.1
## [37] evaluate_0.14 fansi_0.4.2 nlme_3.1-152
## [40] MASS_7.3-54 vegan_2.5-7 tools_4.0.3
## [43] data.table_1.14.0 prettyunits_1.1.1 hms_1.1.0
## [46] lifecycle_1.0.0 stringr_1.4.0 Rhdf5lib_1.12.1
## [49] S4Vectors_0.28.1 munsell_0.5.0 cluster_2.1.2
## [52] Biostrings_2.58.0 ade4_1.7-16 compiler_4.0.3
## [55] jquerylib_0.1.4 rlang_0.4.11 rhdf5_2.34.0
## [58] grid_4.0.3 iterators_1.0.13 rhdf5filters_1.2.1
## [61] biomformat_1.18.0 igraph_1.2.6 labeling_0.4.2
## [64] rmarkdown_2.8 gtable_0.3.0 codetools_0.2-18
## [67] multtest_2.46.0 DBI_1.1.1 reshape2_1.4.4
## [70] R6_2.5.0 knitr_1.33 dplyr_1.0.6
## [73] utf8_1.2.1 fastmatch_1.1-0 permute_0.9-5
## [76] stringi_1.6.2 parallel_4.0.3 Rcpp_1.0.6
## [79] vctrs_0.3.8 tidyselect_1.1.1 xfun_0.23