Single Cell Analysis with Seurat and some custom code!
Seurat is a popular R package that is designed for QC, analysis, and exploration of single cell data. Seurat aims to enable users to identify and interpret sources of heterogeneity from single cell transcriptomic measurements, and to integrate diverse types of single cell data. Further, the authors provide several tutorials on their website.
We start with loading needed libraries for R
library(Seurat)
library(tximport)
library(ggVennDiagram)
Load the Expression Matrix Data and create the combined base Seurat object.
Seurat provides a function Read10X to read in 10X data folder. First we read in data from each individual sample folder. Then, we initialize the Seurat object (CreateSeuratObject) with the raw (non-normalized data). Keep all genes expressed in >= 3 cells. Keep all cells with at least 200 detected genes. Also extracting sample names, calculating and adding in the metadata mitochondrial percentage of each cell. Some QA/QC Finally, saving the raw Seurat object.
A ens2sym.txt file
Reading in the salmon file, we will need to convert the ensembl ids to gene symbols. just like we did [here] (https://ucdavis-bioinformatics-training.github.io/2020-August-Advanced-scRNAseq/data_reduction/scMapping) to create the txp2gene.txt file from biomart, we will want to do the same for the ens2sym.txt file. You will need 3 columns “Gene stable ID”, “Gene stable ID version”, and “Gene name”. Your final file should look like this
Gene stable ID Gene stable ID version Gene name
ENSMUSG00000064372 ENSMUSG00000064372.1 mt-Tp
ENSMUSG00000064371 ENSMUSG00000064371.1 mt-Tt
ENSMUSG00000064370 ENSMUSG00000064370.1 mt-Cytb
ENSMUSG00000064369 ENSMUSG00000064369.1 mt-Te
ENSMUSG00000064368 ENSMUSG00000064368.1 mt-Nd6
ENSMUSG00000064367 ENSMUSG00000064367.1 mt-Nd5
ENSMUSG00000064366 ENSMUSG00000064366.1 mt-Tl2
ENSMUSG00000064365 ENSMUSG00000064365.1 mt-Ts2
ENSMUSG00000064364 ENSMUSG00000064364.1 mt-Th
ENSMUSG00000064363 ENSMUSG00000064363.1 mt-Nd4
ENSMUSG00000065947 ENSMUSG00000065947.3 mt-Nd4l
ENSMUSG00000064361 ENSMUSG00000064361.1 mt-Tr
ENSMUSG00000064360 ENSMUSG00000064360.1 mt-Nd3
## Cellranger
cellranger_orig <- Read10X_h5("Adv_comparison_outputs/654/outs/filtered_feature_bc_matrix.h5")
# If hdf5 isn't working read in from the mtx files
#cellranger_orig <- Read10X("Adv_comparison_outputs/654/outs/filtered_feature_bc_matrix")
s_cellranger_orig <- CreateSeuratObject(counts = cellranger_orig, min.cells = 3, min.features = 200, project = "cellranger")
s_cellranger_orig
An object of class Seurat
15256 features across 4939 samples within 1 assay
Active assay: RNA (15256 features, 0 variable features)
cellranger_htstream <- Read10X_h5("Adv_comparison_outputs/654_htstream/outs/filtered_feature_bc_matrix.h5")
s_cellranger_hts <- CreateSeuratObject(counts = cellranger_htstream, min.cells = 3, min.features = 200, project = "cellranger_hts")
s_cellranger_hts
An object of class Seurat
15252 features across 4933 samples within 1 assay
Active assay: RNA (15252 features, 0 variable features)
## STAR
star <- Read10X("Adv_comparison_outputs/654_htstream_star/outs/filtered_feature_bc_matrix" )
s_star_hts <- CreateSeuratObject(counts = star, min.cells = 3, min.features = 200, project = "star")
s_star_hts
An object of class Seurat
15118 features across 4099 samples within 1 assay
Active assay: RNA (15118 features, 0 variable features)
## SALMON
txi <- tximport("Adv_comparison_outputs/654_htstream_salmon_decoys/alevin/quants_mat.gz", type="alevin")
importing alevin data is much faster after installing `fishpond` (>= 1.2.0)
reading in alevin gene-level counts across cells
## salmon is in ensembl IDs, need to convert to gene symbol
head(rownames(txi$counts))
[1] "ENSMUSG00000064370.1" "ENSMUSG00000064368.1" "ENSMUSG00000064367.1"
[4] "ENSMUSG00000064363.1" "ENSMUSG00000065947.3" "ENSMUSG00000064360.1"
ens2symbol <- read.table("ens2sym.txt",sep="\t",header=T,as.is=T)
map <- ens2symbol$Gene.name[match(rownames(txi$counts),ens2symbol$Gene.stable.ID.version)]
txi_counts <- txi$counts[-which(duplicated(map)),]
map <- map[-which(duplicated(map))]
rownames(txi_counts) <- map
dim(txi_counts)
[1] 35805 3919
s_salmon_hts <- CreateSeuratObject(counts = txi_counts , min.cells = 3, min.features = 200, project = "salmon")
s_salmon_hts
An object of class Seurat
15630 features across 3918 samples within 1 assay
Active assay: RNA (15630 features, 0 variable features)
# Need to Check Rows names/Col names before merge
# they however have different looking cell ids, need to fix
head(colnames(s_cellranger_orig))
[1] "AAACCTGAGATCACGG-1" "AAACCTGAGCATCATC-1" "AAACCTGAGCGCTCCA-1"
[4] "AAACCTGAGTGGGATC-1" "AAACCTGCAGACAGGT-1" "AAACCTGCATCATCCC-1"
head(colnames(s_star_hts))
[1] "AAACCTGAGATCACGG" "AAACCTGAGCATCATC" "AAACCTGAGCGCTCCA" "AAACCTGAGTGGGATC"
[5] "AAACCTGCAGACAGGT" "AAACCTGGTATAGGTA"
head(colnames(s_salmon_hts))
[1] "TGGGAAGCACTACAGT" "CACATAGAGACTAGGC" "GTTTCTAGTTCCACAA" "TACTCATCATAGGATA"
[5] "CACAGGCCAATCTGCA" "GTAGGCCAGGACAGCT"
s_cellranger_orig <- RenameCells(s_cellranger_orig, new.names = sapply(X = strsplit(colnames(s_cellranger_orig), split = "-"), FUN = "[", 1))
s_cellranger_hts <- RenameCells(s_cellranger_hts, new.names = sapply(X = strsplit(colnames(s_cellranger_hts), split = "-"), FUN = "[", 1))
## Merge the dataset
s_merged <- merge(s_cellranger_orig, y = c(s_cellranger_hts, s_star_hts, s_salmon_hts), add.cell.ids = c("cr.orig", "cr.hts", "star.hts", "salmon.hts"), project = "MapTest")
s_merged
An object of class Seurat
17218 features across 17889 samples within 1 assay
Active assay: RNA (17218 features, 0 variable features)
[1] "cr.orig_AAACCTGAGATCACGG" "cr.orig_AAACCTGAGCATCATC"
[3] "cr.orig_AAACCTGAGCGCTCCA" "cr.orig_AAACCTGAGTGGGATC"
[5] "cr.orig_AAACCTGCAGACAGGT" "cr.orig_AAACCTGCATCATCCC"
[1] "salmon.hts_AAAGTAGGTACGAAAT" "salmon.hts_GTGCATAGTAAACACA"
[3] "salmon.hts_CGATGGCCAGGTCTCG" "salmon.hts_ATCCGAAGTGCTGTAT"
[5] "salmon.hts_ACACTGATCGCCCTTA" "salmon.hts_ACACCAAGTGTGACCC"
table(s_merged$orig.ident)
cellranger cellranger_hts salmon star
4939 4933 3918 4099
table(table(sapply(X = strsplit(colnames(s_merged), split = "_"), FUN = "[", 2)))
1 2 3 4
49 735 418 3779
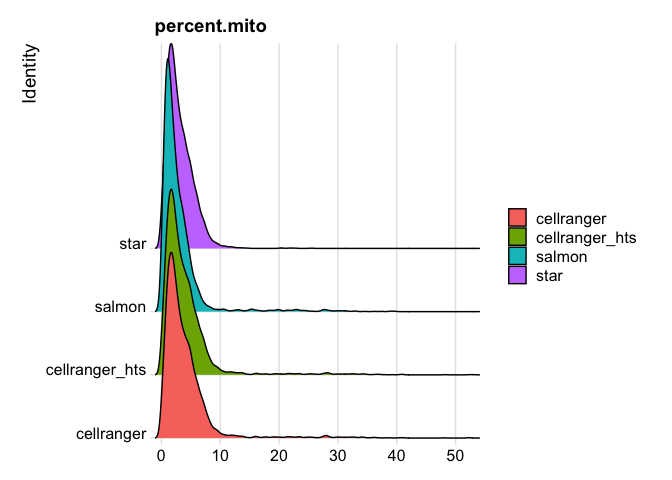
The percentage of reads that map to the mitochondrial genome
- Low-quality / dying cells often exhibit extensive mitochondrial content
- We calculate mitochondrial QC metrics with the PercentageFeatureSet function, which calculates the percentage of counts originating from a set of features.
- We use the set of all genes, in mouse these genes can be identified as those that begin with ‘mt’, in human data they begin with MT.
s_merged$percent.mito <- PercentageFeatureSet(s_merged, pattern = "^mt-")
Lets spend a little time getting to know the Seurat object.
The Seurat object is the center of each single cell analysis. It stores all information associated with the dataset, including data, annotations, analyses, etc. The R function slotNames can be used to view the slot names within an object.
[1] "assays" "meta.data" "active.assay" "active.ident" "graphs"
[6] "neighbors" "reductions" "images" "project.name" "misc"
[11] "version" "commands" "tools"
orig.ident nCount_RNA nFeature_RNA percent.mito
cr.orig_AAACCTGAGATCACGG cellranger 2501 1354 7.3570572
cr.orig_AAACCTGAGCATCATC cellranger 2703 1353 0.8139105
cr.orig_AAACCTGAGCGCTCCA cellranger 2912 1550 3.7087912
cr.orig_AAACCTGAGTGGGATC cellranger 3735 1954 0.5622490
cr.orig_AAACCTGCAGACAGGT cellranger 1123 809 1.2466607
cr.orig_AAACCTGCATCATCCC cellranger 535 428 5.7943925
orig.ident nCount_RNA nFeature_RNA percent.mito
cr.orig_AAACCTGAGATCACGG cellranger 2501 1354 7.3570572
cr.orig_AAACCTGAGCATCATC cellranger 2703 1353 0.8139105
cr.orig_AAACCTGAGCGCTCCA cellranger 2912 1550 3.7087912
cr.orig_AAACCTGAGTGGGATC cellranger 3735 1954 0.5622490
cr.orig_AAACCTGCAGACAGGT cellranger 1123 809 1.2466607
cr.orig_AAACCTGCATCATCCC cellranger 535 428 5.7943925
Question(s)
- What slots are empty, what slots have data?
- What columns are available in meta.data?
- Look up the help documentation for subset?
Now lets do some basic comparisons. Do they share the same cellbarcodes?
ggVennDiagram(list("cr_orig"=colnames(s_cellranger_orig),"cr_hts"=colnames(s_cellranger_hts), "star_hts"=colnames(s_star_hts), "salmon_hts"=colnames(s_salmon_hts)))
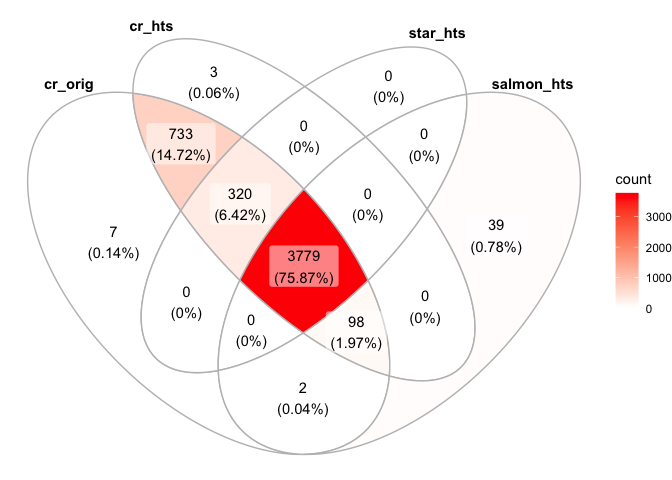
cr_orig_genes <- rowSums(as.matrix(GetAssayData(subset(s_merged, cells=colnames(s_merged)[s_merged$orig.ident=="cellranger"]))))
cr_hts_genes <- rowSums(as.matrix(GetAssayData(subset(s_merged, cells=colnames(s_merged)[s_merged$orig.ident=="cellranger_hts"]))))
star_hts_genes <- rowSums(as.matrix(GetAssayData(subset(s_merged, cells=colnames(s_merged)[s_merged$orig.ident=="star"]))))
salmon_hts_genes <- rowSums(as.matrix(GetAssayData(subset(s_merged, cells=colnames(s_merged)[s_merged$orig.ident=="salmon"]))))
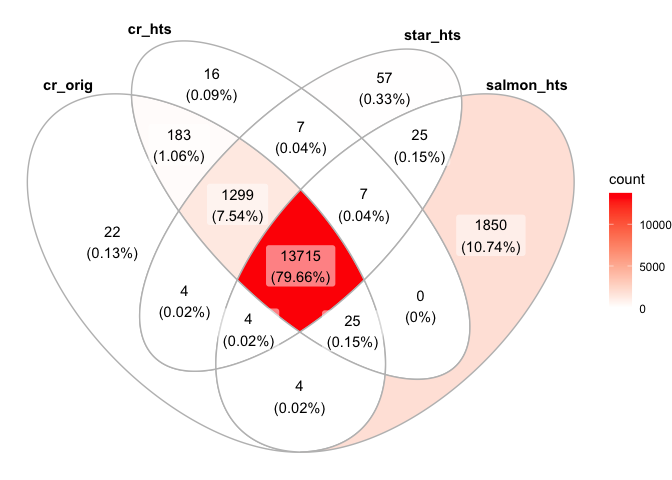
minReads=0
ggVennDiagram(list("cr_orig"=names(cr_orig_genes[cr_orig_genes>minReads]),"cr_hts"=names(cr_hts_genes[cr_hts_genes>minReads]), "star_hts"=names(star_hts_genes[star_hts_genes>minReads]), "salmon_hts"=names(salmon_hts_genes[salmon_hts_genes>minReads])))
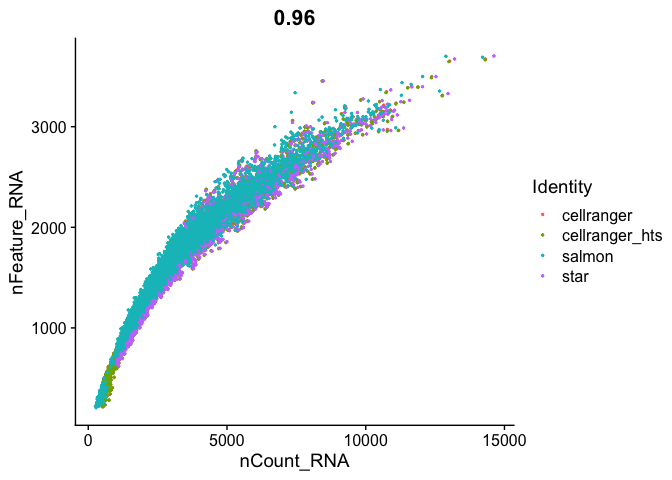
FeatureScatter(
s_merged, "nCount_RNA", "nFeature_RNA",
pt.size = 0.5)
Question(s)
- Spend a minute playing with minReads, see how the data changes.
- What are the sum of UMIs for each?
- Look up the help documentation for subset?
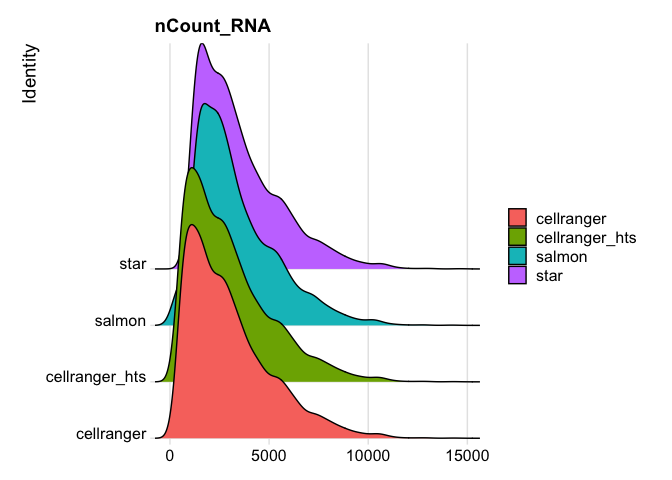
RidgePlot(s_merged, features="nCount_RNA")
Picking joint bandwidth of 338
RidgePlot(s_merged, features="nFeature_RNA")
Picking joint bandwidth of 106
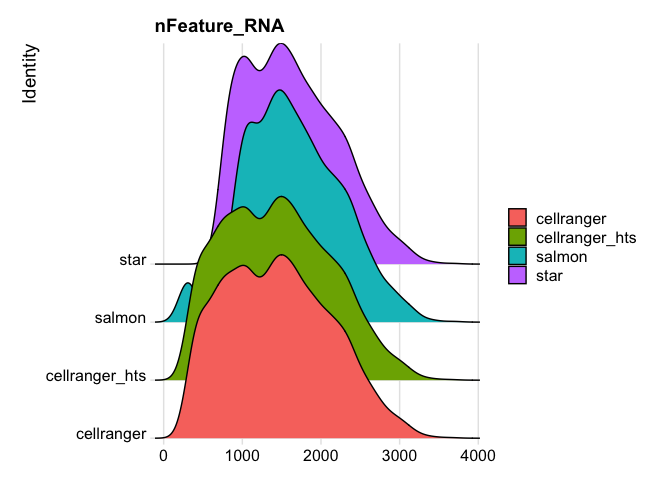
RidgePlot(s_merged, features="percent.mito")
Picking joint bandwidth of 0.367
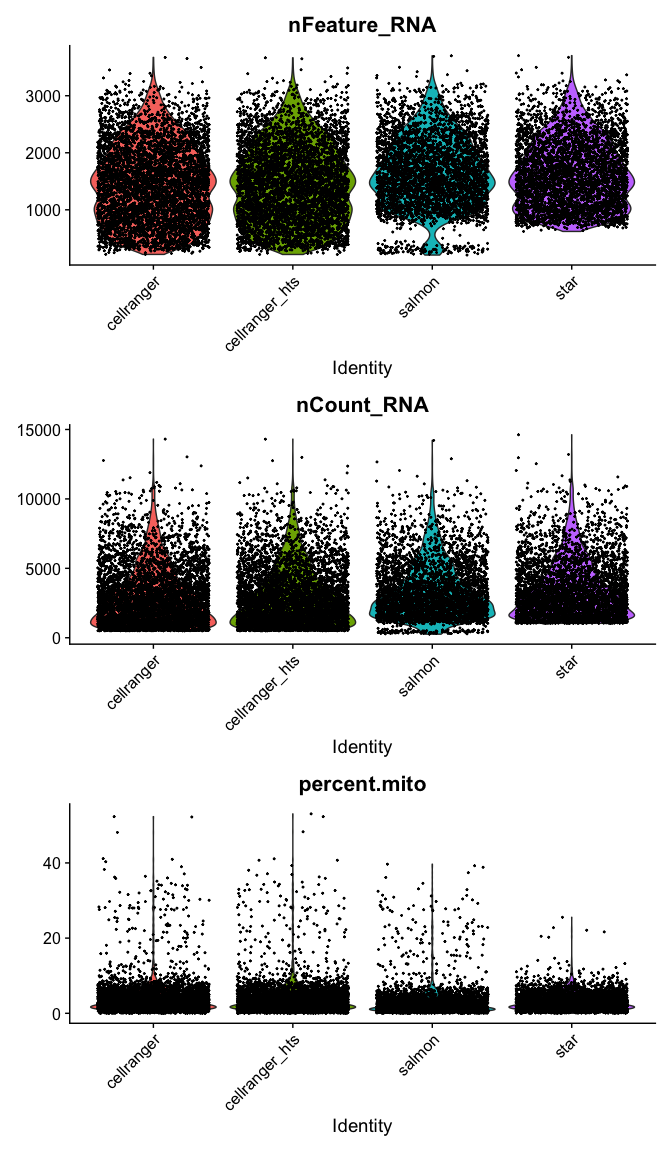
VlnPlot(
s_merged,
features = c("nFeature_RNA", "nCount_RNA","percent.mito"),
ncol = 1, pt.size = 0.3)
s_merged <- NormalizeData(s_merged, normalization.method = "LogNormalize", scale.factor = 10000)
s_merged <- FindVariableFeatures(s_merged, selection.method = "vst", nfeatures = 2000)
all.genes <- rownames(s_merged)
s_merged <- ScaleData(s_merged, features = all.genes)
Centering and scaling data matrix
s_merged <- RunPCA(s_merged, features = VariableFeatures(object = s_merged))
PC_ 1
Positive: Sncg, Ubb, Txn1, Fxyd2, Atp6v0b, Lxn, Sh3bgrl3, Rabac1, Dctn3, Pfn1
Ppia, Fez1, Ndufa11, Cisd1, Ndufa4, Atp6v1f, Atpif1, S100a10, Psmb2, Psmb6
S100a6, Tmem45b, Prdx2, Elob, Nme1, Cox6a1, Gabarapl2, Bex2, Pop5, Bex3
Negative: Adcyap1, Celf4, Gap43, Kit, Atp2b4, Ngfr, Gal, Fxyd7, Lynx1, Spock1
Enah, Meg3, Pcp4, Mt1, Nptn, Eno2, 6330403K07Rik, Syt2, Map2, Unc80
Gpx3, Sv2b, S1pr3, Fgf1, Ptn, Cntn1, Dbpht2, Malat1, S100b, Faim2
PC_ 2
Positive: Cntn1, S100b, Nefh, Thy1, Cplx1, Tagln3, Sv2b, Hopx, Slc17a7, Ntrk3
Lrrn1, Nefm, Atp1b1, Vsnl1, Atp2b2, Nefl, Nat8l, Chchd10, Scn8a, Vamp1
Scn1a, Scn1b, Syt2, Mcam, Endod1, Kcna2, Lynx1, Sh3gl2, Scn4b, Cpne6
Negative: Cd24a, Malat1, Dusp26, Tmem233, Mal2, Prkca, Cd9, Osmr, Tmem158, Nppb
Ift122, Cd44, Carhsp1, Calca, Sst, Tac1, Arpc1b, Npy2r, Gadd45g, Gm525
Gna14, Adcyap1, Bhlhe41, Cd82, Atpaf2, Gpx3, Adk, Scg2, Sntb1, Nts
PC_ 3
Positive: Gm10925, Gm28661, Rps7-ps3, Rpl10-ps3, Gm28437, Gm21981, Gm45234, Rpl9-ps6, Gm20390, Gm21988
Rps27rt, Rpl27-ps3, Gm45713, Gm10177, Gm10288, Gm10175, Kxd1, Rnasek, Gm8430, Rpl10a-ps1
Atp5o, Rpl17-ps3, Nme2, Rps6-ps4, Uba52, Gm12918, Gstp2, Gm14586, Rpl23a-ps3, Rps13-ps2
Negative: Malat1, Aldoa, Meg3, Atp5o.1, D130009I18Rik, D130079A08Rik, AC160336.1, Atpaf2, Gm2694, Nol4l
B230312C02Rik, Ctsl, Ndufb4, Prxl2c, Nip7, Gpx4, Grik1, Synpr, Sult5a1, Tmem245
D930028M14Rik, Gfra2, Gm34455, Zfp202, Ubash3a, Ppp1r1a, Scg3, Mgst3, Gm567, Tubb4b
PC_ 4
Positive: Tspan8, Etv1, Jak1, Tmem233, Resp18, Adk, Nppb, Skp1a, Scg2, Sst
Cystm1, Osmr, Gm525, Nts, Ift122, Nefh, Npy2r, Map7d2, Tesc, Prkca
Nsg1, S100b, Cd82, Calm1, Blvrb, Thy1, Htr1f, Ddah1, Crip2, Carhsp1
Negative: Gm7271, Tafa4, P2ry1, Rarres1, Th, Fxyd6, Wfdc2, Id4, Zfp521, Iqsec2
Gfra2, Rgs5, Tox3, Cdkn1a, Kcnd3, Rgs10, Alcam, Rasgrp1, Rprm, Pou4f2
C1ql4, Piezo2, D130079A08Rik, Synpr, Spink2, Ceacam10, Camk2n1, Bok, Ptpre, Cd81
PC_ 5
Positive: Basp1, Gm765, Cd44, Prkar2b, Calcb, Rab27b, Lpar3, Ctxn3, Calca, Ly86
Mt3, Nefl, Aplp2, Rgs7, Mrgprd, Anks1b, Nrn1l, Nrn1, Gap43, Cd55
Grik1, Tmem255a, Serping1, Rspo2, Klhl5, Nmb, S100a7l2, Synpr, Ptprt, Otoa
Negative: Nppb, Sst, Gm525, Nts, Osmr, Jak1, Htr1f, Npy2r, Hpcal1, Cysltr2
Ptprk, Tesc, Tspan8, Il31ra, Resp18, Blvrb, Etv1, Cmtm7, Cavin1, Ada
Fam178b, Gstt2, Pde4c, Nbl1, Sntb1, Ddah1, Nsg1, Camk2n1, Ptafr, Lgals1
use.pcs = 1:30
s_merged <- FindNeighbors(s_merged, dims = use.pcs)
Computing nearest neighbor graph
Computing SNN
s_merged <- FindClusters(s_merged, resolution = c(0.5,0.75,1.0))
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 17889
Number of edges: 786490
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9490
Number of communities: 28
Elapsed time: 2 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 17889
Number of edges: 786490
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9340
Number of communities: 30
Elapsed time: 2 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 17889
Number of edges: 786490
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9199
Number of communities: 35
Elapsed time: 2 seconds
s_merged <- RunTSNE(s_merged, dims = use.pcs, check_duplicates = FALSE)
s_merged <- RunUMAP(s_merged, dims = use.pcs)
Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per session
07:48:15 UMAP embedding parameters a = 0.9922 b = 1.112
07:48:15 Read 17889 rows and found 30 numeric columns
07:48:15 Using Annoy for neighbor search, n_neighbors = 30
07:48:15 Building Annoy index with metric = cosine, n_trees = 50
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
07:48:17 Writing NN index file to temp file /var/folders/74/h45z17f14l9g34tmffgq9nkw0000gn/T//RtmpYUy6Kh/file25b213101b51
07:48:17 Searching Annoy index using 1 thread, search_k = 3000
07:48:22 Annoy recall = 100%
07:48:22 Commencing smooth kNN distance calibration using 1 thread
07:48:23 Initializing from normalized Laplacian + noise
07:48:24 Commencing optimization for 200 epochs, with 777262 positive edges
07:48:32 Optimization finished
DimPlot(s_merged, reduction = "tsne")
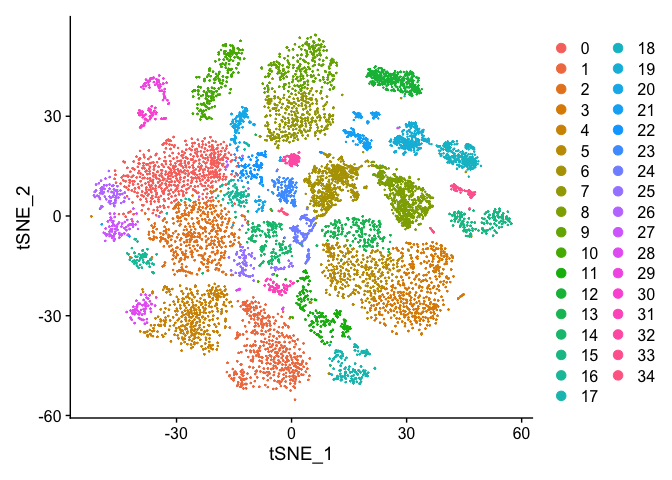
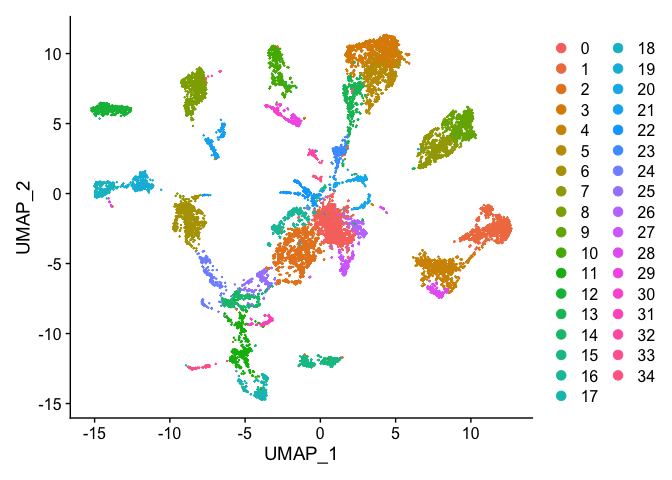
DimPlot(s_merged, reduction = "umap")
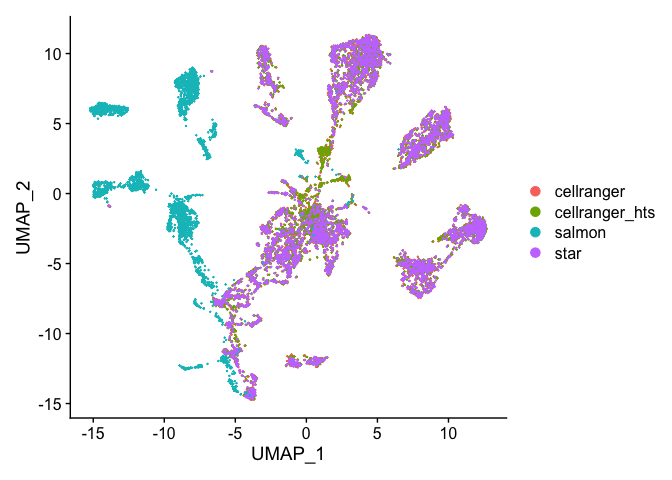
DimPlot(s_merged, group.by = "orig.ident", reduction = "umap")
Finally, save the original object, write out a tab-delimited table that could be read into excel, and view the object.
## Original dataset in Seurat class, with no filtering
save(s_merged,file="mapping_comparison_object.RData")
R version 4.0.2 (2020-06-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] ggVennDiagram_0.3 tximport_1.16.1 Seurat_3.2.0
loaded via a namespace (and not attached):
[1] Rtsne_0.15 colorspace_1.4-1 deldir_0.1-28
[4] ellipsis_0.3.1 class_7.3-17 ggridges_0.5.2
[7] futile.logger_1.4.3 spatstat.data_1.4-3 farver_2.0.3
[10] leiden_0.3.3 listenv_0.8.0 ggrepel_0.8.2
[13] bit64_4.0.2 RSpectra_0.16-0 codetools_0.2-16
[16] splines_4.0.2 knitr_1.29 polyclip_1.10-0
[19] jsonlite_1.7.0 ica_1.0-2 cluster_2.1.0
[22] png_0.1-7 uwot_0.1.8 shiny_1.5.0
[25] sctransform_0.2.1 compiler_4.0.2 httr_1.4.2
[28] Matrix_1.2-18 fastmap_1.0.1 lazyeval_0.2.2
[31] later_1.1.0.1 formatR_1.7 htmltools_0.5.0
[34] tools_4.0.2 rsvd_1.0.3 igraph_1.2.5
[37] gtable_0.3.0 glue_1.4.1 RANN_2.6.1
[40] reshape2_1.4.4 dplyr_1.0.1 Rcpp_1.0.5
[43] spatstat_1.64-1 vctrs_0.3.2 ape_5.4-1
[46] nlme_3.1-148 lmtest_0.9-37 xfun_0.16
[49] stringr_1.4.0 globals_0.12.5 mime_0.9
[52] miniUI_0.1.1.1 lifecycle_0.2.0 irlba_2.3.3
[55] renv_0.11.0 goftest_1.2-2 future_1.18.0
[58] MASS_7.3-51.6 zoo_1.8-8 scales_1.1.1
[61] promises_1.1.1 spatstat.utils_1.17-0 parallel_4.0.2
[64] lambda.r_1.2.4 RColorBrewer_1.1-2 yaml_2.2.1
[67] reticulate_1.16 pbapply_1.4-3 gridExtra_2.3
[70] ggplot2_3.3.2 rpart_4.1-15 stringi_1.4.6
[73] e1071_1.7-3 rlang_0.4.7 pkgconfig_2.0.3
[76] evaluate_0.14 lattice_0.20-41 ROCR_1.0-11
[79] purrr_0.3.4 tensor_1.5 sf_0.9-5
[82] labeling_0.3 patchwork_1.0.1 htmlwidgets_1.5.1
[85] bit_4.0.4 cowplot_1.0.0 tidyselect_1.1.0
[88] RcppAnnoy_0.0.16 plyr_1.8.6 magrittr_1.5
[91] R6_2.4.1 generics_0.0.2 DBI_1.1.0
[94] withr_2.2.0 pillar_1.4.6 mgcv_1.8-31
[97] fitdistrplus_1.1-1 units_0.6-7 survival_3.2-3
[100] abind_1.4-5 tibble_3.0.3 future.apply_1.6.0
[103] crayon_1.3.4 hdf5r_1.3.3 futile.options_1.0.1
[106] KernSmooth_2.23-17 plotly_4.9.2.1 rmarkdown_2.3
[109] grid_4.0.2 data.table_1.13.0 digest_0.6.25
[112] classInt_0.4-3 xtable_1.8-4 VennDiagram_1.6.20
[115] tidyr_1.1.1 httpuv_1.5.4 munsell_0.5.0
[118] viridisLite_0.3.0
