
Velocyto Analysis merging out Seurat analysis with the Velocyto results
We start with loading needed libraries for R
library(Seurat)
library(velocyto.R)
## install SeuratWrappers
#install.packages("devtools")
#library(devtools)
#devtools::install_github('satijalab/seurat-wrappers')
library(SeuratWrappers)
First Download Example Data
download.file("https://bioshare.bioinformatics.ucdavis.edu/bioshare/download/iimg5mz77whzzqc/654.loom", "654.loom")
#RUN Velocity
loom.data <- ReadVelocity(file = "654.loom")
s_cellranger_orig <- as.Seurat(x = loom.data)
Warning: Non-unique features (rownames) present in the input matrix, making
unique
DefaultAssay(object = s_cellranger_orig) <- "spliced"
s_cellranger_orig <- NormalizeData(s_cellranger_orig, verbose = FALSE)
s_cellranger_orig <- FindVariableFeatures(s_cellranger_orig,selection.method = "vst", nfeatures = 2000, verbose = FALSE)
s_cellranger_orig <- ScaleData(s_cellranger_orig, verbose = FALSE)
s_cellranger_orig <- RunPCA(object = s_cellranger_orig, verbose = FALSE)
s_cellranger_orig <- FindNeighbors(object = s_cellranger_orig, dims = 1:30)
Computing nearest neighbor graph
Computing SNN
s_cellranger_orig <- FindClusters(object = s_cellranger_orig)
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4912
Number of edges: 191723
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.9027
Number of communities: 18
Elapsed time: 0 seconds
s_cellranger_orig <- RunUMAP(object = s_cellranger_orig, dims = 1:30)
Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per session
12:26:37 UMAP embedding parameters a = 0.9922 b = 1.112
12:26:37 Read 4912 rows and found 30 numeric columns
12:26:37 Using Annoy for neighbor search, n_neighbors = 30
12:26:37 Building Annoy index with metric = cosine, n_trees = 50
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
12:26:38 Writing NN index file to temp file /var/folders/74/h45z17f14l9g34tmffgq9nkw0000gn/T//RtmpMwEScx/filea0242ed86e75
12:26:38 Searching Annoy index using 1 thread, search_k = 3000
12:26:39 Annoy recall = 100%
12:26:39 Commencing smooth kNN distance calibration using 1 thread
12:26:40 Initializing from normalized Laplacian + noise
12:26:40 Commencing optimization for 500 epochs, with 210556 positive edges
12:26:46 Optimization finished
s_cellranger_orig <- RunVelocity(object = s_cellranger_orig, deltaT = 1, kCells = 25, fit.quantile = 0.02)
Filtering genes in the spliced matrix
Filtering genes in the unspliced matrix
Calculating embedding distance matrix
ident.colors <- (scales::hue_pal())(n = length(x = levels(x = s_cellranger_orig)))
names(x = ident.colors) <- levels(x = s_cellranger_orig)
cell.colors <- ident.colors[Idents(object = s_cellranger_orig)]
names(x = cell.colors) <- colnames(x = s_cellranger_orig)
show.velocity.on.embedding.cor(emb = Embeddings(object = s_cellranger_orig, reduction = "umap"), vel = Tool(object = s_cellranger_orig,
slot = "RunVelocity"), n = 200, scale = "sqrt", cell.colors = ac(x = cell.colors, alpha = 0.5),
cex = 0.8, arrow.scale = 3, show.grid.flow = TRUE, min.grid.cell.mass = 0.5, grid.n = 40, arrow.lwd = 1,
do.par = FALSE, cell.border.alpha = 0.1)
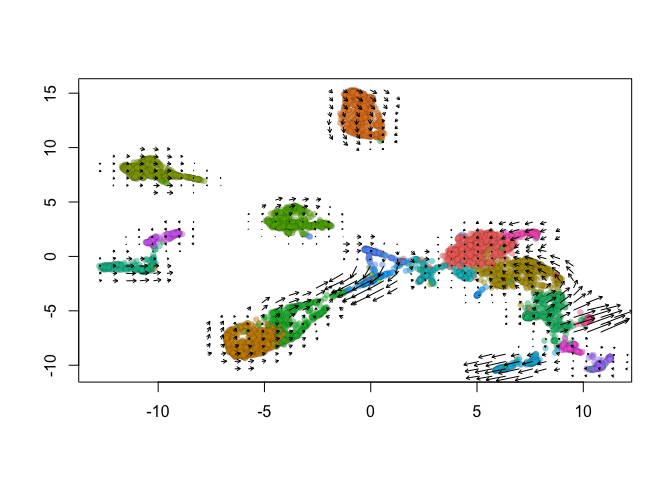
delta projections ... sqrt knn ... transition probs ... done
calculating arrows ... done
grid estimates ... grid.sd= 0.4640639 min.arrow.size= 0.009281278 max.grid.arrow.length= 0.04106169 done
Finally, save the object
## Original dataset in Seurat class, with no filtering
save(s_cellranger_orig,file="Velocyto_object.RData")
Session Information
sessionInfo()
R version 4.0.0 (2020-04-24)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.4
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] SeuratWrappers_0.1.0 velocyto.R_0.6 Matrix_1.2-18
[4] Seurat_3.1.5
loaded via a namespace (and not attached):
[1] nlme_3.1-148 tsne_0.1-3 bit64_0.9-7
[4] RcppAnnoy_0.0.16 RColorBrewer_1.1-2 httr_1.4.1
[7] sctransform_0.2.1 tools_4.0.0 R6_2.4.1
[10] irlba_2.3.3 KernSmooth_2.23-17 uwot_0.1.8
[13] lazyeval_0.2.2 BiocGenerics_0.34.0 mgcv_1.8-31
[16] colorspace_1.4-1 tidyselect_1.1.0 gridExtra_2.3
[19] bit_1.1-15.2 compiler_4.0.0 Biobase_2.48.0
[22] hdf5r_1.3.2 plotly_4.9.2.1 scales_1.1.1
[25] lmtest_0.9-37 ggridges_0.5.2 pbapply_1.4-2
[28] stringr_1.4.0 digest_0.6.25 rmarkdown_2.1
[31] pkgconfig_2.0.3 htmltools_0.4.0 htmlwidgets_1.5.1
[34] rlang_0.4.6 farver_2.0.3 zoo_1.8-8
[37] jsonlite_1.6.1 ica_1.0-2 dplyr_0.8.5
[40] magrittr_1.5 patchwork_1.0.0 Rcpp_1.0.4.6
[43] munsell_0.5.0 ape_5.3 reticulate_1.16
[46] lifecycle_0.2.0 stringi_1.4.6 yaml_2.2.1
[49] MASS_7.3-51.6 Rtsne_0.15 plyr_1.8.6
[52] grid_4.0.0 parallel_4.0.0 listenv_0.8.0
[55] ggrepel_0.8.2 crayon_1.3.4 lattice_0.20-41
[58] cowplot_1.0.0 splines_4.0.0 knitr_1.28
[61] pillar_1.4.4 igraph_1.2.5 future.apply_1.5.0
[64] reshape2_1.4.4 codetools_0.2-16 leiden_0.3.3
[67] glue_1.4.1 evaluate_0.14 pcaMethods_1.80.0
[70] data.table_1.12.8 remotes_2.1.1 renv_0.10.0
[73] BiocManager_1.30.10 png_0.1-7 vctrs_0.3.0
[76] gtable_0.3.0 RANN_2.6.1 purrr_0.3.4
[79] tidyr_1.1.0 future_1.17.0 assertthat_0.2.1
[82] ggplot2_3.3.0 xfun_0.14 rsvd_1.0.3
[85] RSpectra_0.16-0 survival_3.1-12 viridisLite_0.3.0
[88] tibble_3.0.1 cluster_2.1.0 globals_0.12.5
[91] fitdistrplus_1.1-1 ellipsis_0.3.1 ROCR_1.0-11